- Current
- Browse
- Collections
-
For contributors
- For Authors
- Instructions to authors
- Article processing charge
- e-submission
- For Reviewers
- Instructions for reviewers
- How to become a reviewer
- Best reviewers
- For Readers
- Readership
- Subscription
- Permission guidelines
- About
- Editorial policy
Articles
- Page Path
- HOME > Diabetes Metab J > Volume 42(5); 2018 > Article
-
ReviewComplications Pathophysiology of Diabetic Retinopathy: The Old and the New
-
Sentaro Kusuhara1, Yoko Fukushima2, Shuntaro Ogura3,4, Naomi Inoue3, Akiyoshi Uemura3

-
Diabetes & Metabolism Journal 2018;42(5):364-376.
DOI: https://doi.org/10.4093/dmj.2018.0182
Published online: October 22, 2018
1Division of Ophthalmology, Department of Surgery, Kobe University Graduate School of Medicine, Kobe, Japan.
2Department of Ophthalmology, Osaka University Graduate School of Medicine, Osaka, Japan.
3Department of Retinal Vascular Biology, Nagoya City University Graduate School of Medical Sciences, Nagoya, Japan.
4Department of Ophthalmology, Wilmer Ophthalmological Institute, Johns Hopkins Hospital, Baltimore, MD, USA.
- Corresponding author: Akiyoshi Uemura. Department of Retinal Vascular Biology, Nagoya City University Graduate School of Medical Sciences, 1 Kawasumi Mizuho-cho, Mizuho-ku, Nagoya 467-8601, Japan. uemura@med.nagoya-cu.ac.jp
Copyright © 2018 Korean Diabetes Association
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- Vision loss in diabetic retinopathy (DR) is ascribed primarily to retinal vascular abnormalities—including hyperpermeability, hypoperfusion, and neoangiogenesis—that eventually lead to anatomical and functional alterations in retinal neurons and glial cells. Recent advances in retinal imaging systems using optical coherence tomography technologies and pharmacological treatments using anti-vascular endothelial growth factor drugs and corticosteroids have revolutionized the clinical management of DR. However, the cellular and molecular mechanisms underlying the pathophysiology of DR are not fully determined, largely because hyperglycemic animal models only reproduce limited aspects of subclinical and early DR. Conversely, non-diabetic mouse models that represent the hallmark vascular disorders in DR, such as pericyte deficiency and retinal ischemia, have provided clues toward an understanding of the sequential events that are responsible for vision-impairing conditions. In this review, we summarize the clinical manifestations and treatment modalities of DR, discuss current and emerging concepts with regard to the pathophysiology of DR, and introduce perspectives on the development of new drugs, emphasizing the breakdown of the blood-retina barrier and retinal neovascularization.
- Diabetic retinopathy (DR) is the most common microvascular complication in diabetic patients, with a higher incidence in people with type 1 diabetes mellitus compared with type 2 diabetes mellitus [1]. Consistent with the increasing prevalence of diabetes in developed and developing nations, DR is the leading cause of vision loss globally in working middle-aged adults [23]. Based on the presence or absence of retinal neovascularization, DR can be classified clinically into non-proliferative (NPDR) and proliferative (PDR) forms [23]. In eyes with PDR, aberrant neovascularization following retinal ischemia causes vision-threatening vitreous hemorrhage and tractional retinal detachment. Further, diabetic macular edema (DME) affects central vision at any stage of DR. Among diabetic populations, the estimated prevalence of any form of DR is 34.6% (93 million worldwide), and those of PDR and DME are 6.96% and 6.81%, respectively [1].
- A major risk factor for DR is sustained hyperglycemia, but hypertension, dyslipidemia, and pregnancy have also been implicated [123]. Notably, certain diabetic populations do not develop DR despite having these systemic risk factors, whereas good glycemic control might not necessarily eliminate the lifetime risk of DR [2]. These patterns indicate that additional factors, such as genetic susceptibility, are involved in the initiation and progression of DR. Thus, it is often difficult to predict the risk of DR in individual diabetic patients.
- In the past decade, pharmacological therapies using anti-vascular endothelial growth factor (VEGF) drugs and corticosteroids have dramatically changed the clinical management of DR [23]. However, because of their limited efficacy and potential adverse effects, a comprehensive understanding of the pathophysiology of DR is urgently needed for the development of new drugs. In this review, we summarize the current knowledge and emerging concepts of the pathophysiology of DR that have been obtained from the clinic and basic research and introduce perspectives on the development of new drugs.
INTRODUCTION
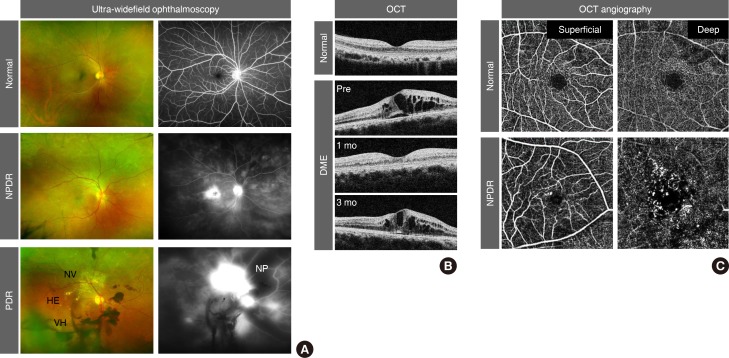
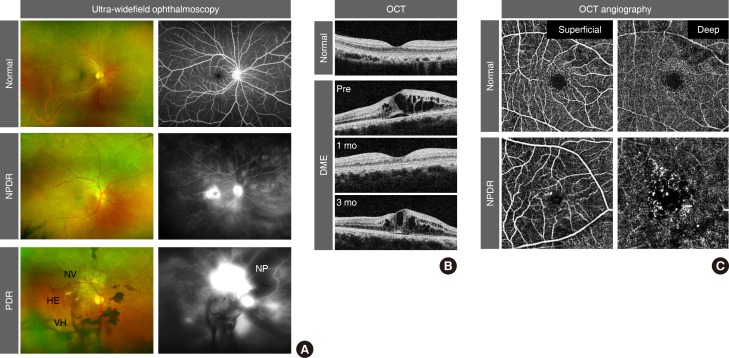
- Because an early diagnosis of DR is crucial for preventing vision loss, routine ophthalmological examinations are recommended for all diabetic patients at severity-dependent intervals [23]. DR can be diagnosed ophthalmoscopically, based on retinal vascular lesions, such as microaneurysms, dot and blot hemorrhages, and deposition of exudative lipoproteins (hard exudates). Fluorescein angiography (FA), in conjunction with ultra-widefield scanning laser ophthalmoscopy, can reveal vascular leakage, non-perfusion, and neovascularization over the entire retina in DR (Fig. 1A). Optical coherence tomography (OCT) generates cross-sectional retinal images, enabling longitudinal assessments of the macular morphology and thickness in eyes with DME (Fig. 1B). In contrast to the potential risk of allergic reactions with FA, OCT angiography (OCTA) noninvasively generates high-resolution images of superficial and deep retinal vascular networks (Fig. 1C). Adaptive optics scanning laser ophthalmoscopy can detect retinal hemorheological changes and cone photoreceptor irregularities in diabetic eyes [45]. Overall and local retinal functions can be evaluated by full-field and multifocal electroretinography (ERG), respectively [6]. These multimodal data might be able to be integrated by artificial intelligence-based systems in the future management of DR [7].
- Non-proliferative diabetic retinopathy
- Based on the severity of retinal vascular lesions, NPDR is categorized into mild, moderate, and severe forms (Table 1) [23]. Whereas mild NPDR exhibits only microaneurysms, moderate NPDR presents with additional signs of impaired vessel integrity and vessel occlusion, including dot and blot hemorrhages, hard exudates, and cotton wool spots. Severe NPDR is accompanied by more distinct features of retinal ischemia, such as venous beading and intra-retinal microvascular abnormalities (IRMAs) that are adjacent to non-perfusion areas.
- For patients with mild to moderate NPDR, systemic control of hyperglycemia, hypertension, and dyslipidemia is critical in preventing the progression and reversing the severity of retinopathy [23]. However, if blood glucose levels decrease rapidly, the DR worsens in 10% to 20% of patients within 3 to 6 months [8]. For severe NPDR, panretinal photocoagulation (PRP) is considered for ablating ischemic neurons and glial cells in non-perfusion areas, thereby reducing their oxygen demand and production of pro-angiogenic growth factors, including VEGF [23]. Although PRP reduces the risk of progression to PDR, its destructive properties can cause peripheral visual field defects and reduced night vision [9]. Moreover, PRP often deteriorates central vision by exacerbating DME, which can be suppressed by adjunct sub-Tenon injections of triamcinolone acetonide, a potent long-acting corticosteroid [10].
- Diabetic macular edema
- By slit-lamp biomicroscopy or OCT, DME can be detected as retinal thickening in the macular areas, which is a consequence of the accumulation of fluid within neural tissues [111213]. Since the 1980s, DME had long been treated initially with focal lasers that targeted leaky microaneurysms and grid lasers that targeted macular areas of diffuse leakage and capillary non-perfusion [1415]. Today, intravitreal anti-VEGF therapy has become the standard of care for DME, based on a series of randomized controlled clinical trials that demonstrated its superiority in improving vision compared with laser therapy [23].
- Bevacizumab (Avastin; Genentech, San Francisco, CA, USA), a humanized monoclonal antibody against VEGFA, was approved by the U.S. Food and Drug Administration (FDA) for metastatic colorectal cancer in 2004 and has been used off-label for the treatment of DME [16]. Subsequently, intravitreal injections of ranibizumab (Lucentis; Genentech), the Fab fragment of a monoclonal anti-VEGFA, and aflibercept (Eylea; Regeneron, Tarrytown, NY, USA), a recombinant VEGF receptor (VEGFR) protein that neutralizes VEGFA, VEGFB, and placental growth factor (PlGF), were approved by the FDA for DME in 2012 and 2014, respectively [16]. The biological properties of VEGF signals are described below. Based on their anti-leakage and anti-angiogenic potency, intravitreal ranibizumab and aflibercept are used globally for DME, age-related macular degeneration, myopic choroidal neovascularization, and macular edema secondary to retinal vein occlusion, whereas off-label intravitreal bevacizumab is administered regionally because of its cost-effectiveness [1617]. In most cases, repeated intravitreal injections of these anti-VEGF agents are needed because of the recurrence of DME (Fig. 1B), which raises concerns over infectious endophthalmitis, cerebro-cardiovascular events, and a greater economic burden [18].
- Intravitreal or sub-Tenon injections of triamcinolone acetonide and intravitreal implants of dexamethasone (Ozurdex; Allergan, Dublin, Ireland) and fluocinolone acetonide (Iluvien, Alimera Sciences, Alpharetta, GA, USA; and Retisert, Bausch & Lomb, Bridgewater, NJ, USA) are also used for DME, although the potential adverse effects of these corticosteroids, including cataract progression and elevations in intraocular pressure, should be monitored carefully [219]. Notably, corticosteroids are often effective for DME refractory to anti-VEGF therapies [19]. Based on their prolonged efficacy and cost-effectiveness, corticosteroids can be a useful option for DME, especially in eyes that have been implanted with intraocular lenses. In cases that experience unsuccessful outcomes with these pharmacological therapies, focal or grid laser remains an alternative therapy. Otherwise, vitrectomy surgery can be considered, particularly for DME that is associated with vitreomacular traction [20].
- Proliferative diabetic retinopathy
- In eyes with PDR, new blood vessels that protrude from the ischemic retinal surface cause vitreous hemorrhages (Fig. 1A) [21]. In approximately 8% of PDR patients, the formation of contractile fibrovascular membranes accompanies aberrant neoangiogenesis, inducing tractional retinal detachment [22]. Persistent retinal hypoxia further leads to neovascularization of the iris and refractory glaucoma [21]. To avoid these devastating consequences, PRP should be applied immediately outside of the macular area [9]. For PDR eyes with sustained vitreous hemorrhage or tractional retinal detachment, vitrectomy should be performed in a timely manner [2].
- Notably, repeated anti-VEGF injections for DME suppress the progression to PDR and mitigate the severity of PDR [2324]. Moreover, repeated anti-VEGF injections for PDR result in better visual acuity and lower rates of vitreous hemorrhage, retinal detachment, and neovascular glaucoma compared with PRP [25262728]. These findings might prompt a shift in the clinical management of PDR, wherein treatment regimens that combine PRP, anti-VEGF agents, and corticosteroids should be optimized, depending on the ocular, systemic, and economic status of individual patients.
CLINICAL MANAGEMENT OF DIABETIC RETINOPATHY
- To gain insights into the cellular and molecular mechanisms that underlie the pathophysiology of DR, diabetic mouse models are frequently employed because of their low maintenance cost and short reproductive cycle and the availability of genetically modified strains [29]. The structure and function of mouse retinas can be monitored longitudinally by ultra-widefield scanning laser ophthalmoscopy, OCT, OCTA, and ERG [293031]. Moreover, 2-photon and confocal laser scanning fluorescence microscopy, combined with a cataract-preventing contact lens in anesthetized mice, enables the in vivo imaging of retinal cell dynamics [32].
- Type 1 diabetes mellitus mice, induced by β-cell destruction with streptozotocin (STZ) or by a spontaneous dominant-negative mutation in the insulin-2 gene (Akita mouse), recapitulate several features of early DR, including hyperpermeability and degeneration of retinal vessels [29]. However, these mice fail to reproduce any signs of advanced DR [29]. As alternative DR models, non-diabetic mice that overexpress or lack specific genes have been developed. For example, a transgenic mouse line that overexpresses insulin-like growth factor-1 develops retinal non-perfusion, IRMA, and neovascularization [33]. In addition, overexpression of VEGF and hyperglycemia in Akimba mice synergistically enhances the vascular abnormalities that are characteristic of DR [29]. With the recent advances in genomic engineering with CRISPR-Cas9 technology [34], mutant mouse models will expand our understanding of the causative roles of specific molecules in the pathophysiology of DR.
- Hyperglycemia, oxidative stress, and inflammation
- The metabolic abnormalities of diabetes induce the overproduction of mitochondrial superoxide in vascular endothelial cells (ECs), which subsequently leads to increased flux through the polyol pathway, the production of advanced glycation end-products (AGEs), upregulation of the receptor for AGEs and its activating ligands, activation of the protein kinase C pathway, and overactivity of the hexosamine pathway [35]. These pathways elevate the levels of intracellular reactive oxygen species and cause irreversible cell damage through epigenetic changes, such as histone modifications, DNA methylation, and non-coding RNAs [3536]. Consistent with this concept of “metabolic/hyperglycemic memory,” euglycemic re-entry after transplantation of pancreatic islet cells to STZ-induced diabetic mice fails to heal retinal microvascular damage [37]. These findings might explain the effects of early glycemic control on the future development of DR [38].
- Under sustained hyperglycemia, oxidative stress, various signaling pathways, and epigenetic modifications induce inflammation (Fig. 2) [353639]. The levels of pro-inflammatory cytokines and chemokines, such as monocyte chemoattractant protein 1 (MCP-1), tumor necrosis factor α (TNF-α), interleukin 1β (IL-1β), and IL-6, are elevated in eyes with DR [40]. The pivotal functions of inflammation in the initiation and progression of DR have been corroborated empirically with the therapeutic efficacy of corticosteroids for DME and DR per se [41]. In diabetic retinas, the adhesion and infiltration of leukocytes might damage vascular ECs and neuroglial cells by physical occlusion of capillaries and through the release of inflammatory mediators and superoxide [42]. Thus, novel anti-inflammatory drugs with fewer adverse effects than corticosteroids are desired for the treatment of DR. To date, several compounds and antibody drugs that target inflammatory signals, such as MCP-1, TNF-α, IL-1β, and IL-6, have been evaluated clinically for DME or DR [1940], but none of them has been approved.
- Although it remains unknown why retinal microvascular abnormalities develop over years of hyperglycemic periods (more than 5 years in type 2 diabetes mellitus), clinical and experimental evidence has demonstrated irreversible loss of neurons preceding vascular lesions in diabetic retinas (Fig. 2) [43444546]. Thus, neuroprotective agents, such as eye drops of somatostatin or brimonidine (an α-2 adrenergic receptor agonist), are expected to prevent neuroglial degeneration and preserve long-term vision in subclinical and early DR (EUROCONDOR study [NCT01726075]) [4748].
- Vascular endothelial growth factors
- In 1948, Michaelson postulated the presence of a pro-angiogenic factor derived from hypoxic retinas in DR [49]. After the discovery of VEGF in the 1980s [5051], increased VEGF levels were reported in eyes with PDR in 1994 [52]. Then, VEGF injections into monkey eyes reproduced the retinal vascular abnormalities that were seen in NPDR and PDR [53]. Subsequently, extensive research on the physiological and pathological functions of VEGF led to the development of anti-VEGF drugs [16].
- The VEGF family, comprising VEGFA, VEGFB, VEGFC, VEGFD, and PlGF, are secretory glycoprotein ligands, each of which binds distinctly to the transmembrane tyrosine kinases VEGFR1, VEGFR2, and VEGFR3 (Fig. 2) [5455]. VEGFA (herein referred to as VEGF unless otherwise noted) binds to VEGFR1 and VEGFR2, whereas VEGFB and PlGF bind only to VEGFR1 [5455]. VEGFC and VEGFD bind to VEGFR3 and regulate lymphangiogenesis, whereas proteolytic processing of these ligands allows binding to VEGFR2 [5455].
- During hypoxia, VEGFA is upregulated transcriptionally, and alternative mRNA splicing generates several VEGFA isoforms, such as VEGFA121, VEGFA165, and VEGFA189, in human [545556]. These VEGFA isoforms have disparate binding affinities to extracellular matrices and a VEGFR2 co-receptor, neuropilin-1. Post-translational processing of VEGFA proteins further diversifies their distribution and signaling activities [54]. In vascular ECs, the binding of VEGFA to VEGFR2 activates several signal transduction cascades, including the mitogen-activated protein kinase pathway and the phosphatidyl inositol 3-kinase (PI3K)/Akt pathway, thereby promoting cell proliferation and migration and the subsequent formation of new blood vessels [5455]. Further, the VEGFA-VEGFR2 signal disrupts EC-EC adherens and tight junctions, leading to vascular hyperpermeability and fluid extravasation [5455]. Thus, the VEGFA-VEGFR2 signal is pivotal in retinal angiogenesis and vascular leakage in DR (Fig. 2) [4956].
- In ECs, transmembrane and soluble VEGFR1 functions as a decoy for VEGFA, modulating the intensity of the VEGFR2 signal [5455]. The physiological functions of endothelial VEGFR1 signaling, activated by VEGFA, VEGFB, or PlGF, are assumed to be negligible, because mice that lack the intracellular kinase domain of VEGFR1 are viable and have no vascular abnormalities [57]. Conversely, the VEGFR1 signal in monocytes and macrophages contributes significantly under inflammatory conditions [58], correlating with the upregulation of VEGFB and PlGF in eyes with DR [59].
- In contrast to its deleterious functions in pathological settings, VEGFA has been implicated in the maintenance of the homeostasis of neural retinas, in which a subset of neurons and Müller glia constitutively express VEGFR2 [60]. Furthermore, VEGFA that is secreted from retinal pigment epithelium (RPE) cells is indispensable for maintaining choroidal vessels [6162], raising concerns over the harmful effects of repeat anti-VEGF injections. Nonetheless, new anti-VEGF drugs are still being developed to prolong the potency and injection intervals in the treatment of DR. Among them, brolucizumab (RTH258), a humanized single-chain antibody fragment against VEGFA, is expected to reduce the injection frequency because of its small molecular weight and high intravitreal concentration [63]. Currently, the KITE (NCT03481660) and KESTREL (NCT03481634) phase 3 clinical trials are evaluating the efficacy and safety of brolucizumab for DME compared with aflibercept.
- Angiopoietins
- Angiopoietin-1 (Ang1) was identified as an agonistic ligand of endothelial tyrosine kinase with immunoglobulin-like loops and epidermal growth factor homology domains 2 (Tie2) receptor in 1996 by Regeneron Pharmaceuticals Inc. [64]. The binding of Ang1 to Tie2 activates the PI3K/Akt pathway, leading to the phosphorylation and inactivation of a forkhead box transcription factor, forkhead box O1 (FOXO1), in ECs (Fig. 2). This signaling stabilizes vessel integrity by promoting EC survival, preventing vascular permeability, and suppressing inflammatory responses [65]. In 1997, Regeneron reported another ligand, Ang2, that binds Tie2 with similar affinity as Ang1 [66]. However, Ang2 weakly activates Tie2 in ECs. Thus, Ang2 was assumed to be a natural Tie2 antagonist that counteracts Ang1-mediated vessel stabilization.
- Ang2 renders ECs more sensitive to pro-angiogenic, pro-permeable, and pro-inflammatory stimuli, such as VEGFA and TNF-α [65]. Moreover, Ang2 is upregulated by hypoxia, VEGFA, and hyperglycemia [6768], whereas Ang2-induced activation of FOXO1 upregulates Ang2, forming a positive feedback loop [65]. These findings indicate that Ang2 facilitates angiogenesis, vascular permeability, and inflammation under certain disease settings. Ang2 is upregulated in eyes with DR, age-related macular degeneration, and retinal vein occlusion [6970].
- To date, a series of Ang2 blockers and Tie2 activators have been developed [71]. Among them, the Tie2 activator AKB-9778 [72], the anti-Ang2/VEGFA bispecific antibody RG7716 [69], and the fully human monoclonal anti-Ang2 nesvacumab (REGN910) [73] have been evaluated clinically for treating DR. The phase 2 RUBY study (NCT02712008) for DME reported no further improvement with the combination of aflibercept and nesvacumab compared with aflibercept alone. On the other hand, the phase 2 TIME-2 study (NCT02050828) of the combination of AKB-9778 and ranibizumab and the phase 2 BOULEVARD study (NCT02699450) of RG7716 reported favorable outcomes in the treatment of DME [74]. Notably, Ang2 can act as a Tie2 agonist, depending on the environment [7576]. Thus, anti-Ang2 drugs might inhibit the agonistic activity of Ang2 and result in unexpected outcomes.
- Breakdown of the blood-retina barrier
- To maintain retinal homeostasis, the leakage of plasma into neural tissues is regulated tightly by the inner and outer blood-retina barrier (BRB), sealed by retinal vascular ECs and RPE cells, respectively [111213]. Although dysfunction of the RPE can increase the influx of fluid from the underlying choroidal vessels in diabetic eyes, the pathogenic role of the breakdown of the outer BRB in DME is not fully understood [1112]. Conversely, elevated paracellular and transcellular leakage in retinal ECs causes the inner BRB to break down in DR [111213]. Based on the seminal histopathological observations of human diabetic eyes [77], the consensus is that pericyte dropout from retinal capillary walls is responsible for breakdown of the inner BRB (Fig. 2).
- Retinal pericytes originate from the neural crest and regulate blood flow by providing mechanical strength to the vessel walls [78]. In addition, pericytes are pivotal in maintaining EC integrity via secretory signals and direct cell-cell contact [78]. In developing retinas, EC-derived platelet-derived growth factor (PDGF) B promotes the recruitment of PDGF receptor (PDGFR) β-expressing pericytes to nascent blood vessels [78]. Therefore, disruptions in the PDGFB-PDGFRβ signal in postnatal mice can deplete pericytes from growing retinal vessels, leading to vessel enlargement, hyperpermeability, hypoperfusion, retinal edema, and hemorrhage [798081]. Notably, transient inhibition of pericyte recruitment during development results in persistent EC-pericyte dissociation in adult retinas, with vascular lesions that are characteristic of DR [81]. In pericyte-deficient retinas, the ECs of superficial retinal vessels proliferate actively but fail to migrate down into the deeper layers, forming aneurysm-like structures with excess accumulation of ECs [81]. In human DR, mitotic ECs are also found in microaneurysms [77], whereas microaneurysms occasionally disappear after intravitreal anti-VEGF injection [82]. These findings suggest that the formation of microaneurysms in DR is in part attributed to the over-proliferation of pericyte-deficient ECs in response to VEGFA.
- In mouse retinas, a deficiency in pericytes induces endothelial inflammation and perivascular macrophage infiltration [8081]. In this setting, macrophage-derived VEGFA activates endothelial VEGFR2, whereas VEGFA and PlGF activate VEGFR1 in macrophages in an autocrine manner. Moreover, pericyte-free ECs upregulate Ang2 and undergo FOXO1 nuclear translocation, especially in microaneurysms, forming an Ang2-FOXO1-based positive feedback loop [8081]. These experimental results indicate that pericyte deficiency in growing retinal vessels elicits a cycle of damage due to EC-macrophage interactions, leading to sustained inflammation and irreversible breakdown of the BRB [83]. Unexpectedly, however, stripping pericytes from adult retinas sensitizes ECs to VEGFA but is insufficient to induce alterations in vessel structure and function [80]. Moreover, the PDGFB-PDGFRβ signal is dispensable for maintaining EC-pericyte associations in adult retinas [80]. Thus, further investigation is required to determine the causes and consequences of pericyte dropout in the pathophysiology of DR.
- Retinal neovascularization
- During development, intra-retinal growth of new blood vessels delivers oxygen to neural tissues efficiently [8485], in contrast to extra-retinal vascular outgrowth, which fails to resolve the tissue hypoxia in PDR (Fig. 2). Comparative analyses of physiological and pathological angiogenesis in mouse retinas have provided mechanistic insights into vessel guidance. In postnatal mouse retinas, the ECs of developing blood vessels migrate over the extracellular matrix scaffolds that are formed by the preexisting astrocyte network [848586]. Retinal astrocytes further establish the concentration gradients of matrix-binding VEGFA that is secreted by them or by neurons, promoting intra-retinal projections of endothelial filopodia from the sprouting vascular tips [87]. Concurrently, chemorepulsive signals, such as neuron-derived semaphorin 3E (Sema3E), which binds to endothelial PlexinD1 receptor, retract disoriented endothelial filopodia, thereby rectifying angiogenic directions [8889].
- Conversely, in an oxygen-induced retinopathy (OIR) mouse model, retinal ischemia that follows vessel regression under hyperoxia (75% O2 from postnatal day 7 to 12) evokes centripetal vascular regrowth, which gives rise to extra-retinal vascular tufts [90]. In this setting, degenerative astrocytes fail to form extracellular fibronectin matrices, whereas retinal neurons, but not astrocytes, predominantly express VEGFA [88]. Thus, defective physical scaffolds for EC migration and disrupted spatial distribution of VEGFA proteins may be responsible for the extra-retinal neoangiogenesis. Notably, the ECs of extra-retinal vessels prominently express PlexinD1, and intravitreal Sema3E injections selectively suppress disoriented angiogenesis without affecting retinal vascular regeneration in the OIR model [88]. Given the decreased levels of aqueous Sema3E in human eyes with PDR [91], supplementation with intravitreal Sema3E should have clinical benefit in preventing aberrant neoangiogenesis. To facilitate vascular regeneration in ischemic retinas, new modalities that restore the pro-angiogenic activities of retinal astrocytes should be developed.
- In the formation of fibrovascular membranes that are associated with extra-retinal neoangiogenesis, a series of pro-fibrotic signals, such as transforming growth factor β, PDGF, and connective tissue growth factor, have been implicated in the transdifferentiation, proliferation, and migration of myofibroblasts and their production of contractile matrix [2292]. Nevertheless, the origins of retinal myofibroblasts remain unknown in PDR [22]. Conversely, cell-fate mapping analyses in mouse models of fibrosis have demonstrated the potential of pericytes and perivascular mesenchymal cells to transdifferentiate into myofibroblasts in various tissues and organs [93]. Given the rapid development or progression of tractional retinal detachment after injections of anti-VEGF drugs into PDR eyes [94], it is postulated that the remaining pericytes after EC ablation from retinal neovascularization constitute a source of myofibroblasts, which should be validated experimentally in future studies.
PATHOPHYSIOLOGY OF DIABETIC RETINOPATHY
- Complementary clinical and experimental evidence has increased our understanding of the pathophysiology of DR. However, the molecular backgrounds that are responsible for retinal vascular abnormalities may vary in individual eyes with DR, as evidenced by their differential responses to anti-VEGF drugs and corticosteroids. Moreover, a substantial question remains unanswere d as to why the retina is preferentially affected in diabetic patients. To determine the common and retina-specific mechanisms underlying diabetic microvascular complications, the broad areas of biomedical research, including ophthalmology, diabetology, neuroscience, immunology, and vascular biology, will need to be integrated. These efforts will optimize personalized medicine by combining drugs with distinct modes of action in the future treatment of DR.
CONCLUSIONS
-
Acknowledgements
- We thank all previous and current members of the Uemura laboratory who contributed to the original studies cited in this review manuscript. This work was supported in part by grants from the Japan Society for the Promotion of Science KAKENHI (16H05155), the Japan Science and Technology Agency CREST “Spontaneous pattern formation ex vivo,” and the Takeda Science Foundation to Akiyoshi Uemura. We thank Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript.
ACKNOWLEDGMENTS
-
CONFLICT OF INTERESTS: No potential conflict of interest relevant to this article was reported.
NOTES
- 1. Yau JW, Rogers SL, Kawasaki R, Lamoureux EL, Kowalski JW, Bek T, Chen SJ, Dekker JM, Fletcher A, Grauslund J, Haffner S, Hamman RF, Ikram MK, Kayama T, Klein BE, Klein R, Krishnaiah S, Mayurasakorn K, O'Hare JP, Orchard TJ, Porta M, Rema M, Roy MS, Sharma T, Shaw J, Taylor H, Tielsch JM, Varma R, Wang JJ, Wang N, West S, Xu L, Yasuda M, Zhang X, Mitchell P, Wong TY. Meta-Analysis for Eye Disease (META-EYE) Study Group. Global prevalence and major risk factors of diabetic retinopathy. Diabetes Care 2012;35:556-564. ArticlePubMedPMCPDF
- 2. Wong TY, Sun J, Kawasaki R, Ruamviboonsuk P, Gupta N, Lansingh VC, Maia M, Mathenge W, Moreker S, Muqit MMK, Resnikoff S, Verdaguer J, Zhao P, Ferris F, Aiello LP, Taylor HR. Guidelines on diabetic eye care: the International Council of Ophthalmology Recommendations for screening, follow-up, referral, and treatment based on resource settings. Ophthalmology 2018;125:1608-1622. ArticlePubMed
- 3. Solomon SD, Chew E, Duh EJ, Sobrin L, Sun JK, VanderBeek BL, Wykoff CC, Gardner TW. Diabetic retinopathy: a position statement by the American Diabetes Association. Diabetes Care 2017;40:412-418. ArticlePubMedPMCPDF
- 4. Arichika S, Uji A, Murakami T, Unoki N, Yoshitake S, Dodo Y, Ooto S, Miyamoto K, Yoshimura N. Retinal hemorheologic characterization of early-stage diabetic retinopathy using adaptive optics scanning laser ophthalmoscopy. Invest Ophthalmol Vis Sci 2014;55:8513-8522. ArticlePubMed
- 5. Lammer J, Prager SG, Cheney MC, Ahmed A, Radwan SH, Burns SA, Silva PS, Sun JK. Cone photoreceptor irregularity on adaptive optics scanning laser ophthalmoscopy correlates with severity of diabetic retinopathy and macular edema. Invest Ophthalmol Vis Sci 2016;57:6624-6632. ArticlePubMedPMC
- 6. Bearse MA Jr, Adams AJ, Han Y, Schneck ME, Ng J, Bronson-Castain K, Barez S. A multifocal electroretinogram model predicting the development of diabetic retinopathy. Prog Retin Eye Res 2006;25:425-448. ArticlePubMedPMC
- 7. Schmidt-Erfurth U, Sadeghipour A, Gerendas BS, Waldstein SM, Bogunovic H. Artificial intelligence in retina. Prog Retin Eye Res 2018 8 01 [Epub]. Article
- 8. Feldman-Billard S, Larger E, Massin P. Standards for screeningand surveillance of ocular complications in people with diabetes SFD study group. Early worsening of diabetic retinopathy after rapid improvement of blood glucose control in patients with diabetes. Diabetes Metab 2018;44:4-14. ArticlePubMed
- 9. Bressler NM, Beck RW, Ferris FL 3rd. Panretinal photocoagulation for proliferative diabetic retinopathy. N Engl J Med 2011;365:1520-1526. ArticlePubMed
- 10. Unoki N, Nishijima K, Kita M, Suzuma K, Watanabe D, Oh H, Kimura T, Sakamoto A, Yoshimura N. Randomised controlled trial of posterior sub-Tenon triamcinolone as adjunct to panretinal photocoagulation for treatment of diabetic retinopathy. Br J Ophthalmol 2009;93:765-770. ArticlePubMed
- 11. Das A, McGuire PG, Rangasamy S. Diabetic macular edema: pathophysiology and novel therapeutic targets. Ophthalmology 2015;122:1375-1394. ArticlePubMed
- 12. Daruich A, Matet A, Moulin A, Kowalczuk L, Nicolas M, Sellam A, Rothschild PR, Omri S, Gelize E, Jonet L, Delaunay K, De Kozak Y, Berdugo M, Zhao M, Crisanti P, Behar-Cohen F. Mechanisms of macular edema: beyond the surface. Prog Retin Eye Res 2018;63:20-68. ArticlePubMed
- 13. Klaassen I, Van Noorden CJ, Schlingemann RO. Molecular basis of the inner blood-retinal barrier and its breakdown in diabetic macular edema and other pathological conditions. Prog Retin Eye Res 2013;34:19-48. ArticlePubMed
- 14. Early Treatment Diabetic Retinopathy Study report number 1. Early Treatment Diabetic Retinopathy Study research group. Photocoagulation for diabetic macular edema. Arch Ophthalmol 1985;103:1796-1806. ArticlePubMed
- 15. Early Treatment Diabetic Retinopathy Study Report Number 2. Early Treatment Diabetic Retinopathy Study Research Group. Treatment techniques and clinical guidelines for photocoagulation of diabetic macular edema. Ophthalmology 1987;94:761-774. ArticlePubMed
- 16. Ferrara N, Adamis AP. Ten years of anti-vascular endothelial growth factor therapy. Nat Rev Drug Discov 2016;15:385-403. ArticlePubMedPDF
- 17. van Asten F, Michels CTJ, Hoyng CB, van der Wilt GJ, Klevering BJ, Rovers MM, Grutters JPC. The cost-effectiveness of bevacizumab, ranibizumab and aflibercept for the treatment of age-related macular degeneration: a cost-effectiveness analysis from a societal perspective. PLoS One 2018;13:e0197670. ArticlePubMedPMC
- 18. Lally DR, Shah CP, Heier JS. Vascular endothelial growth factor and diabetic macular edema. Surv Ophthalmol 2016;61:759-768. ArticlePubMed
- 19. Bolinger MT, Antonetti DA. Moving past anti-VEGF: novel therapies for treating diabetic retinopathy. Int J Mol Sci 2016;17:E1498Article
- 20. Diabetic Retinopathy Clinical Research Network Writing Committee. Haller JA, Qin H, Apte RS, Beck RR, Bressler NM, Browning DJ, Danis RP, Glassman AR, Googe JM, Kollman C, Lauer AK, Peters MA, Stockman ME. Vitrectomy outcomes in eyes with diabetic macular edema and vitreomacular traction. Ophthalmology 2010;117:1087-1093. ArticlePubMed
- 21. Cheung N, Mitchell P, Wong TY. Diabetic retinopathy. Lancet 2010;376:124-136. ArticlePubMed
- 22. Shu DY, Lovicu FJ. Myofibroblast transdifferentiation: the dark force in ocular wound healing and fibrosis. Prog Retin Eye Res 2017;60:44-65. ArticlePubMedPMC
- 23. Nguyen QD, Brown DM, Marcus DM, Boyer DS, Patel S, Feiner L, Gibson A, Sy J, Rundle AC, Hopkins JJ, Rubio RG, Ehrlich JS. RISE and RIDE Research Group. Ranibizumab for diabetic macular edema: results from 2 phase III randomized trials: RISE and RIDE. Ophthalmology 2012;119:789-801. ArticlePubMed
- 24. Mitchell P, McAllister I, Larsen M, Staurenghi G, Korobelnik JF, Boyer DS, Do DV, Brown DM, Katz TA, Berliner A, Vitti R, Zeitz O, Metzig C, Lu C, Holz FG. Evaluating the impact of intravitreal aflibercept on diabetic retinopathy progression in the VIVID-DME and VISTA-DME studies. Ophthalmol Retina 2018;2:988-996.ArticlePubMed
- 25. Krick TW, Bressler NM. Recent clinically relevant highlights from the Diabetic Retinopathy Clinical Research Network. Curr Opin Ophthalmol 2018;29:199-205. ArticlePubMed
- 26. Writing Committee for the Diabetic Retinopathy Clinical Research Network. Gross JG, Glassman AR, Jampol LM, Inusah S, Aiello LP, Antoszyk AN, Baker CW, Berger BB, Bressler NM, Browning D, Elman MJ, Ferris FL 3rd, Friedman SM, Marcus DM, Melia M, Stockdale CR, Sun JK, Beck RW. Panretinal photocoagulation vs intravitreous ranibizumab for proliferative diabetic retinopathy: a randomized clinical trial. JAMA 2015;314:2137-2146. ArticlePubMedPMC
- 27. Sivaprasad S, Prevost AT, Vasconcelos JC, Riddell A, Murphy C, Kelly J, Bainbridge J, Tudor-Edwards R, Hopkins D, Hykin P. CLARITY Study Group. Clinical efficacy of intravitreal aflibercept versus panretinal photocoagulation for best corrected visual acuity in patients with proliferative diabetic retinopathy at 52 weeks (CLARITY): a multicentre, single-blinded, randomised, controlled, phase 2b, non-inferiority trial. Lancet 2017;389:2193-2203. PubMed
- 28. Bressler SB, Beaulieu WT, Glassman AR, Gross JG, Jampol LM, Melia M, Peters MA, Rauser ME. Diabetic Retinopathy Clinical Research Network. Factors associated with worsening proliferative diabetic retinopathy in eyes treated with panretinal photocoagulation or ranibizumab. Ophthalmology 2017;124:431-439. ArticlePubMed
- 29. Robinson R, Barathi VA, Chaurasia SS, Wong TY, Kern TS. Update on animal models of diabetic retinopathy: from molecular approaches to mice and higher mammals. Dis Model Mech 2012;5:444-456. ArticlePubMedPMCPDF
- 30. Nakao S, Arita R, Nakama T, Yoshikawa H, Yoshida S, Enaida H, Hafezi-Moghadam A, Matsui T, Ishibashi T. Wide-field laser ophthalmoscopy for mice: a novel evaluation system for retinal/choroidal angiogenesis in mice. Invest Ophthalmol Vis Sci 2013;54:5288-5293. ArticlePubMed
- 31. Park JR, Choi W, Hong HK, Kim Y, Jun Park S, Hwang Y, Kim P, Woo SJ, Park KH, Oh WY. Imaging laser-induced choroidal neovascularization in the rodent retina using optical coherence tomography angiography. Invest Ophthalmol Vis Sci 2016;57:OCT331-OCT340. ArticlePubMed
- 32. Ikeda W, Nakatani T, Uemura A. Cataract-preventing contact lens for in vivo imaging of mouse retina. Biotechniques 2018;65:101-104. ArticlePubMed
- 33. Ruberte J, Ayuso E, Navarro M, Carretero A, Nacher V, Haurigot V, George M, Llombart C, Casellas A, Costa C, Bosch A, Bosch F. Increased ocular levels of IGF-1 in transgenic mice lead to diabetes-like eye disease. J Clin Invest 2004;113:1149-1157. ArticlePubMedPMC
- 34. Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 2013;8:2281-2308. ArticlePubMedPMCPDF
- 35. Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res 2010;107:1058-1070. ArticlePubMedPMC
- 36. Reddy MA, Zhang E, Natarajan R. Epigenetic mechanisms in diabetic complications and metabolic memory. Diabetologia 2015;58:443-455. ArticlePubMedPDF
- 37. Friedrichs P, Schlotterer A, Sticht C, Kolibabka M, Wohlfart P, Dietrich A, Linn T, Molema G, Hammes HP. Hyperglycaemic memory affects the neurovascular unit of the retina in a diabetic mouse model. Diabetologia 2017;60:1354-1358. ArticlePubMedPDF
- 38. Aiello LP. DCCT/EDIC Research Group. Diabetic retinopathy and other ocular findings in the diabetes control and complications trial/epidemiology of diabetes interventions and complications study. Diabetes Care 2014;37:17-23. ArticlePubMedPDF
- 39. El-Osta A, Brasacchio D, Yao D, Pocai A, Jones PL, Roeder RG, Cooper ME, Brownlee M. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J Exp Med 2008;205:2409-2417. ArticlePubMedPMCPDF
- 40. Rubsam A, Parikh S, Fort PE. Role of inflammation in diabetic retinopathy. Int J Mol Sci 2018;19:E942
- 41. Wykoff CC. Impact of intravitreal pharmacotherapies including antivascular endothelial growth factor and corticosteroid agents on diabetic retinopathy. Curr Opin Ophthalmol 2017;28:213-218. ArticlePubMed
- 42. Miyamoto K, Khosrof S, Bursell SE, Rohan R, Murata T, Clermont AC, Aiello LP, Ogura Y, Adamis AP. Prevention of leukostasis and vascular leakage in streptozotocin-induced diabetic retinopathy via intercellular adhesion molecule-1 inhibition. Proc Natl Acad Sci U S A 1999;96:10836-10841. ArticlePubMedPMC
- 43. Sohn EH, van Dijk HW, Jiao C, Kok PH, Jeong W, Demirkaya N, Garmager A, Wit F, Kucukevcilioglu M, van Velthoven ME, DeVries JH, Mullins RF, Kuehn MH, Schlingemann RO, Sonka M, Verbraak FD, Abramoff MD. Retinal neurodegeneration may precede microvascular changes characteristic of diabetic retinopathy in diabetes mellitus. Proc Natl Acad Sci U S A 2016;113:E2655-E2664. ArticlePubMedPMC
- 44. van Dijk HW, Kok PH, Garvin M, Sonka M, Devries JH, Michels RP, van Velthoven ME, Schlingemann RO, Verbraak FD, Abramoff MD. Selective loss of inner retinal layer thickness in type 1 diabetic patients with minimal diabetic retinopathy. Invest Ophthalmol Vis Sci 2009;50:3404-3409. ArticlePubMed
- 45. Santos AR, Ribeiro L, Bandello F, Lattanzio R, Egan C, Frydkjaer-Olsen U, Garcia-Arumi J, Gibson J, Grauslund J, Harding SP, Lang GE, Massin P, Midena E, Scanlon P, Aldington SJ, Simao S, Schwartz C, Ponsati B, Porta M, Costa MA, Hernandez C, Cunha-Vaz J, Simo R. European Consortium for the Early Treatment of Diabetic Retinopathy (EUROCONDOR). Functional and structural findings of neurodegeneration in early stages of diabetic retinopathy: cross-sectional analyses of baseline data of the EUROCONDOR project. Diabetes 2017;66:2503-2510. ArticlePubMedPDF
- 46. Barber AJ, Lieth E, Khin SA, Antonetti DA, Buchanan AG, Gardner TW. Neural apoptosis in the retina during experimental and human diabetes. Early onset and effect of insulin. J Clin Invest 1998;102:783-791. ArticlePubMedPMC
- 47. Simo R, Hernandez C. European Consortium for the Early Treatment of Diabetic Retinopathy (EUROCONDOR). Neurodegeneration in the diabetic eye: new insights and therapeutic perspectives. Trends Endocrinol Metab 2014;25:23-33. ArticlePubMed
- 48. Barber AJ, Baccouche B. Neurodegeneration in diabetic retinopathy: potential for novel therapies. Vision Res 2017;139:82-92. ArticlePubMed
- 49. Campochiaro PA, Aiello LP, Rosenfeld PJ. Anti-vascular endothelial growth factor agents in the treatment of retinal disease: from bench to bedside. Ophthalmology 2016;123:S78-S88. ArticlePubMed
- 50. Senger DR, Galli SJ, Dvorak AM, Perruzzi CA, Harvey VS, Dvorak HF. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science 1983;219:983-985. ArticlePubMed
- 51. Leung DW, Cachianes G, Kuang WJ, Goeddel DV, Ferrara N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 1989;246:1306-1309. ArticlePubMed
- 52. Aiello LP, Avery RL, Arrigg PG, Keyt BA, Jampel HD, Shah ST, Pasquale LR, Thieme H, Iwamoto MA, Park JE, Nguyen HV, Aiello LM, Ferrara N, King GL. Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N Engl J Med 1994;331:1480-1487. ArticlePubMed
- 53. Tolentino MJ, Miller JW, Gragoudas ES, Jakobiec FA, Flynn E, Chatzistefanou K, Ferrara N, Adamis AP. Intravitreous injections of vascular endothelial growth factor produce retinal ischemia and microangiopathy in an adult primate. Ophthalmology 1996;103:1820-1828. ArticlePubMed
- 54. Simons M, Gordon E, Claesson-Welsh L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat Rev Mol Cell Biol 2016;17:611-625. ArticlePubMedPDF
- 55. Koch S, Claesson-Welsh L. Signal transduction by vascular endothelial growth factor receptors. Cold Spring Harb Perspect Med 2012;2:a006502ArticlePubMedPMC
- 56. Miller JW, Le Couter J, Strauss EC, Ferrara N. Vascular endothelial growth factor a in intraocular vascular disease. Ophthalmology 2013;120:106-114. ArticlePubMed
- 57. Hiratsuka S, Minowa O, Kuno J, Noda T, Shibuya M. Flt-1 lacking the tyrosine kinase domain is sufficient for normal development and angiogenesis in mice. Proc Natl Acad Sci U S A 1998;95:9349-9354. ArticlePubMedPMC
- 58. Murakami M, Iwai S, Hiratsuka S, Yamauchi M, Nakamura K, Iwakura Y, Shibuya M. Signaling of vascular endothelial growth factor receptor-1 tyrosine kinase promotes rheumatoid arthritis through activation of monocytes/macrophages. Blood 2006;108:1849-1856. ArticlePubMedPDF
- 59. Mesquita J, Castro-de-Sousa JP, Vaz-Pereira S, Neves A, Passarinha LA, Tomaz CT. Vascular endothelial growth factors and placenta growth factor in retinal vasculopathies: current research and future perspectives. Cytokine Growth Factor Rev 2018;39:102-115. ArticlePubMed
- 60. Saint-Geniez M, Maharaj AS, Walshe TE, Tucker BA, Sekiyama E, Kurihara T, Darland DC, Young MJ, D'Amore PA. Endogenous VEGF is required for visual function: evidence for a survival role on muller cells and photoreceptors. PLoS One 2008;3:e3554. ArticlePubMedPMC
- 61. Saint-Geniez M, Kurihara T, Sekiyama E, Maldonado AE, D'Amore PA. An essential role for RPE-derived soluble VEGF in the maintenance of the choriocapillaris. Proc Natl Acad Sci U S A 2009;106:18751-18756. ArticlePubMedPMC
- 62. Kurihara T, Westenskow PD, Bravo S, Aguilar E, Friedlander M. Targeted deletion of VEGFA in adult mice induces vision loss. J Clin Invest 2012;122:4213-4217. ArticlePubMedPMC
- 63. Holz FG, Dugel PU, Weissgerber G, Hamilton R, Silva R, Bandello F, Larsen M, Weichselberger A, Wenzel A, Schmidt A, Escher D, Sararols L, Souied E. Single-chain antibody fragment VEGF inhibitor rth258 for neovascular age-related macular degeneration: a randomized controlled study. Ophthalmology 2016;123:1080-1089. ArticlePubMed
- 64. Davis S, Aldrich TH, Jones PF, Acheson A, Compton DL, Jain V, Ryan TE, Bruno J, Radziejewski C, Maisonpierre PC, Yancopoulos GD. Isolation of angiopoietin-1, a ligand for the TIE2 receptor, by secretion-trap expression cloning. Cell 1996;87:1161-1169. ArticlePubMed
- 65. Augustin HG, Koh GY, Thurston G, Alitalo K. Control of vascular morphogenesis and homeostasis through the angiopoietin-Tie system. Nat Rev Mol Cell Biol 2009;10:165-177. ArticlePubMedPDF
- 66. Maisonpierre PC, Suri C, Jones PF, Bartunkova S, Wiegand SJ, Radziejewski C, Compton D, McClain J, Aldrich TH, Papadopoulos N, Daly TJ, Davis S, Sato TN, Yancopoulos GD. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science 1997;277:55-60. ArticlePubMed
- 67. Oh H, Takagi H, Suzuma K, Otani A, Matsumura M, Honda Y. Hypoxia and vascular endothelial growth factor selectively up-regulate angiopoietin-2 in bovine microvascular endothelial cells. J Biol Chem 1999;274:15732-15739. ArticlePubMed
- 68. Yao D, Taguchi T, Matsumura T, Pestell R, Edelstein D, Giardino I, Suske G, Rabbani N, Thornalley PJ, Sarthy VP, Hammes HP, Brownlee M. High glucose increases angiopoietin-2 transcription in microvascular endothelial cells through methylglyoxal modification of mSin3A. J Biol Chem 2007;282:31038-31045. ArticlePubMed
- 69. Regula JT, Lundh von Leithner P, Foxton R, Barathi VA, Cheung CM, Bo Tun SB, Wey YS, Iwata D, Dostalek M, Moelleken J, Stubenrauch KG, Nogoceke E, Widmer G, Strassburger P, Koss MJ, Klein C, Shima DT, Hartmann G. Targeting key angiogenic pathways with a bispecific CrossMAb optimized for neovascular eye diseases. EMBO Mol Med 2016;8:1265-1288. ArticlePubMedPMCPDF
- 70. Watanabe D, Suzuma K, Suzuma I, Ohashi H, Ojima T, Kurimoto M, Murakami T, Kimura T, Takagi H. Vitreous levels of angiopoietin 2 and vascular endothelial growth factor in patients with proliferative diabetic retinopathy. Am J Ophthalmol 2005;139:476-481. ArticlePubMed
- 71. Saharinen P, Eklund L, Alitalo K. Therapeutic targeting of the angiopoietin-TIE pathway. Nat Rev Drug Discov 2017;16:635-661. ArticlePubMedPDF
- 72. Shen J, Frye M, Lee BL, Reinardy JL, McClung JM, Ding K, Kojima M, Xia H, Seidel C, Lima e Silva R, Dong A, Hackett SF, Wang J, Howard BW, Vestweber D, Kontos CD, Peters KG, Campochiaro PA. Targeting VE-PTP activates TIE2 and stabilizes the ocular vasculature. J Clin Invest 2014;124:4564-4576. ArticlePubMedPMC
- 73. Papadopoulos KP, Kelley RK, Tolcher AW, Razak AR, Van Loon K, Patnaik A, Bedard PL, Alfaro AA, Beeram M, Adriaens L, Brownstein CM, Lowy I, Kostic A, Trail PA, Gao B, DiCioccio AT, Siu LL. A phase I first-in-human study of nesvacumab (REGN910), a fully human anti-angiopoietin-2 (Ang2) monoclonal antibody, in patients with advanced solid tumors. Clin Cancer Res 2016;22:1348-1355. ArticlePubMedPDF
- 74. Campochiaro PA, Khanani A, Singer M, Patel S, Boyer D, Dugel P, Kherani S, Withers B, Gambino L, Peters K, Brigell M. TIME-2 Study Group. Enhanced benefit in diabetic macular edema from AKB-9778 Tie2 activation combined with vascular endothelial growth factor suppression. Ophthalmology 2016;123:1722-1730. ArticlePubMed
- 75. Korhonen EA, Lampinen A, Giri H, Anisimov A, Kim M, Allen B, Fang S, D'Amico G, Sipila TJ, Lohela M, Strandin T, Vaheri A, Ylä-Herttuala S, Koh GY, McDonald DM, Alitalo K, Saharinen P. Tie1 controls angiopoietin function in vascular remodeling and inflammation. J Clin Invest 2016;126:3495-3510. ArticlePubMedPMC
- 76. Kim M, Allen B, Korhonen EA, Nitschke M, Yang HW, Baluk P, Saharinen P, Alitalo K, Daly C, Thurston G, McDonald DM. Opposing actions of angiopoietin-2 on Tie2 signaling and FOXO1 activation. J Clin Invest 2016;126:3511-3525. ArticlePubMedPMC
- 77. Cogan DG, Toussaint D, Kuwabara T. Retinal vascular patterns. IV. Diabetic retinopathy. Arch Ophthalmol 1961;66:366-378. ArticlePubMed
- 78. Armulik A, Genove G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell 2011;21:193-215. ArticlePubMed
- 79. Uemura A, Ogawa M, Hirashima M, Fujiwara T, Koyama S, Takagi H, Honda Y, Wiegand SJ, Yancopoulos GD, Nishikawa S. Recombinant angiopoietin-1 restores higher-order architecture of growing blood vessels in mice in the absence of mural cells. J Clin Invest 2002;110:1619-1628. ArticlePubMedPMC
- 80. Park DY, Lee J, Kim J, Kim K, Hong S, Han S, Kubota Y, Augustin HG, Ding L, Kim JW, Kim H, He Y, Adams RH, Koh GY. Plastic roles of pericytes in the blood-retinal barrier. Nat Commun 2017;8:15296ArticlePubMedPMCPDF
- 81. Ogura S, Kurata K, Hattori Y, Takase H, Ishiguro-Oonuma T, Hwang Y, Ahn S, Park I, Ikeda W, Kusuhara S, Fukushima Y, Nara H, Sakai H, Fujiwara T, Matsushita J, Ema M, Hirashima M, Minami T, Shibuya M, Takakura N, Kim P, Miyata T, Ogura Y, Uemura A. Sustained inflammation after pericyte depletion induces irreversible blood-retina barrier breakdown. JCI Insight 2017;2:e90905. ArticlePubMedPMC
- 82. Ehlers JP, Wang K, Singh RP, Babiuch AS, Schachat AP, Yuan A, Reese JL, Stiegel L, Srivastava SK. A prospective randomized comparative dosing trial of ranibizumab in bevacizumab-resistant diabetic macular edema: the REACT study. Ophthalmol Retina 2018;2:217-224. PubMedPMC
- 83. Uemura A. Pharmacologic management of diabetic retinopathy. J Biochem 2018;163:3-9. ArticlePubMed
- 84. Selvam S, Kumar T, Fruttiger M. Retinal vasculature development in health and disease. Prog Retin Eye Res 2018;63:1-19. ArticlePubMed
- 85. Uemura A, Kusuhara S, Katsuta H, Nishikawa S. Angiogenesis in the mouse retina: a model system for experimental manipulation. Exp Cell Res 2006;312:676-683. ArticlePubMed
- 86. Uemura A, Kusuhara S, Wiegand SJ, Yu RT, Nishikawa S. Tlx acts as a proangiogenic switch by regulating extracellular assembly of fibronectin matrices in retinal astrocytes. J Clin Invest 2006;116:369-377. ArticlePubMedPMC
- 87. Gerhardt H, Golding M, Fruttiger M, Ruhrberg C, Lundkvist A, Abramsson A, Jeltsch M, Mitchell C, Alitalo K, Shima D, Betsholtz C. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J Cell Biol 2003;161:1163-1177. ArticlePubMedPMCPDF
- 88. Fukushima Y, Okada M, Kataoka H, Hirashima M, Yoshida Y, Mann F, Gomi F, Nishida K, Nishikawa S, Uemura A. Sema3E-PlexinD1 signaling selectively suppresses disoriented angiogenesis in ischemic retinopathy in mice. J Clin Invest 2011;121:1974-1985. ArticlePubMedPMC
- 89. Kim J, Oh WJ, Gaiano N, Yoshida Y, Gu C. Semaphorin 3E-Plexin-D1 signaling regulates VEGF function in developmental angiogenesis via a feedback mechanism. Genes Dev 2011;25:1399-1411. ArticlePubMedPMC
- 90. Smith LE, Wesolowski E, McLellan A, Kostyk SK, D'Amato R, Sullivan R, D'Amore PA. Oxygen-induced retinopathy in the mouse. Invest Ophthalmol Vis Sci 1994;35:101-111. PubMed
- 91. Kwon SH, Shin JP, Kim IT, Park DH. Aqueous levels of angiopoietin-like 4 and semaphorin 3E correlate with nonperfusion area and macular volume in diabetic retinopathy. Ophthalmology 2015;122:968-975. ArticlePubMed
- 92. Klaassen I, van Geest RJ, Kuiper EJ, van Noorden CJ, Schlingemann RO. The role of CTGF in diabetic retinopathy. Exp Eye Res 2015;133:37-48. ArticlePubMed
- 93. Di Carlo SE, Peduto L. The perivascular origin of pathological fibroblasts. J Clin Invest 2018;128:54-63. ArticlePubMedPMC
- 94. Arevalo JF, Maia M, Flynn HW Jr, Saravia M, Avery RL, Wu L, Eid Farah M, Pieramici DJ, Berrocal MH, Sanchez JG. Tractional retinal detachment following intravitreal bevacizumab (Avastin) in patients with severe proliferative diabetic retinopathy. Br J Ophthalmol 2008;92:213-216. ArticlePubMed
REFERENCES
Clinical features of diabetic retinopathy (DR). (A) Pseudo-colored fundus (left) and fluorescein angiography (right) images from ultra-widefield ophthalmoscopy. Note the elevated leakage of fluorescein dye in the macular area in non-proliferative DR (NPDR) and from aberrant neovascularization (NV) in proliferative DR (PDR). Dark areas in fluorescein angiography represent vascular non-perfusion (NP). (B) Cross-sectional macular images from optical coherence tomography (OCT). Note the recurrence of diabetic macular edema (DME) at 3 months after intravitreal anti-vascular endothelial growth factor injection. (C) Superficial and deep retinal vessel images from OCT angiography. Note the microaneurysms and enlargement of the foveal avascular zone in NPDR. HE, hard exudate; VH, vitreous hemorrhage.
Schematic of key cellular and molecular events in the progression of diabetic retinopathy. Hyperglycemia initiates oxidative stress, epigenetic modifications, and inflammation in vascular endothelial cells (ECs). Neuroglial degeneration precedes microvascular changes. Pericyte loss from vessel walls sensitizes ECs to microenvironmental stimuli. Infiltrating macrophages secrete vascular endothelial growth factor (VEGF) A and placental growth factor (PlGF). A positive feedback loop between angiopoietin-2 (Ang2) and a forkhead box transcription factor, forkhead Box O1 (FOXO1), in ECs further destabilizes vessel integrity. These events form a cycle of vessel damage, leading to the breakdown of the blood-retina barrier. Retinal hypoxia resulting from vessel occlusion induces extra-retinal neoangiogenesis accompanied by fibrovascular membrane formation. Throughout these processes, signal transduction via the mitogen-activated protein kinase (MAPK) and the phosphatidyl inositol 3-kinase (PI3K)/Akt pathways downstream of VEGF receptor (VEGFR) 2 in ECs is pivotal in retinal angiogenesis and vascular leakage. Tie2, tyrosine kinase with immunoglobulin-like loops and epidermal growth factor homology domains 2.

Classification of diabetic retinopathy and recommended eye care
DR, diabetic retinopathy; NPDR, non-proliferative DR; IRMA, intra-retinal microvascular abnormality; PDR, proliferative DR; PRP, panretinal photocoagulation; DME, diabetic macular edema; VEGF, vascular endothelial growth factor.
aIntravitreal ranibizumab is approved by the U.S. Food and Drug Administration to treat all forms of DR, with or without DME.
Figure & Data
References
Citations
- Recent Insights into the Etiopathogenesis of Diabetic Retinopathy and Its Management
Arpon Biswas, Abhijit Deb Choudhury, Sristi Agrawal, Amol Chhatrapati Bisen, Sachin Nashik Sanap, Sarvesh Kumar Verma, Mukesh Kumar, Anjali Mishra, Shivansh Kumar, Mridula Chauhan, Rabi Sankar Bhatta
Journal of Ocular Pharmacology and Therapeutics.2024; 40(1): 13. CrossRef - GMFB/AKT/TGF‐β3 in Müller cells mediated early retinal degeneration in a streptozotocin‐induced rat diabetes model
Tong Zhu, Yingao Li, Lilin Zhu, Jinyuan Xu, Zijun Feng, Hao Chen, Si Shi, Caiying Liu, Qingjian Ou, Furong Gao, Jieping Zhang, Caixia Jin, Jingying Xu, Jiao Li, Jingfa Zhang, Yanlong Bi, Guo‐tong Xu, Juan Wang, Haibin Tian, Lixia Lu
Glia.2024; 72(3): 504. CrossRef - The significance of glutaredoxins for diabetes mellitus and its complications
Mengmeng Zhou, Eva-Maria Hanschmann, Axel Römer, Thomas Linn, Sebastian Friedrich Petry
Redox Biology.2024; 71: 103043. CrossRef - Proteomic analysis of diabetic retinopathy identifies potential plasma-protein biomarkers for diagnosis and prognosis
Bent Honoré, Javad Nouri Hajari, Tobias Torp Pedersen, Tomas Ilginis, Hajer Ahmad Al-Abaiji, Claes Sepstrup Lønkvist, Jon Peiter Saunte, Dorte Aalund Olsen, Ivan Brandslund, Henrik Vorum, Carina Slidsborg
Clinical Chemistry and Laboratory Medicine (CCLM).2024;[Epub] CrossRef - Next generation therapeutics for retinal neurodegenerative diseases
Matthew B. Appell, Jahnavi Pejavar, Ashwin Pasupathy, Sri Vishnu Kiran Rompicharla, Saed Abbasi, Kiersten Malmberg, Patricia Kolodziejski, Laura M. Ensign
Journal of Controlled Release.2024; 367: 708. CrossRef - Modeling early pathophysiological phenotypes of diabetic retinopathy in a human inner blood-retinal barrier-on-a-chip
Thomas L. Maurissen, Alena J. Spielmann, Gabriella Schellenberg, Marc Bickle, Jose Ricardo Vieira, Si Ying Lai, Georgios Pavlou, Sascha Fauser, Peter D. Westenskow, Roger D. Kamm, Héloïse Ragelle
Nature Communications.2024;[Epub] CrossRef - Emerging role of ferroptosis in diabetic retinopathy: a review
Ruohong Wang, Suyun Rao, Zheng Zhong, Ke Xiao, Xuhui Chen, Xufang Sun
Journal of Drug Targeting.2024; 32(4): 393. CrossRef - Serum vitamin D is substantially reduced and predicts flares in diabetic retinopathy patients
Yong Zhuang, Zihao Zhuang, Qingyan Cai, Xin Hu, Huibin Huang
Journal of Diabetes Investigation.2024;[Epub] CrossRef - Ocular pharmacological and biochemical profiles of 6-thioguanine: a drug repurposing study
Maria Consiglia Trotta, Carlo Gesualdo, Caterina Claudia Lepre, Marina Russo, Franca Ferraraccio, Iacopo Panarese, Ernesto Marano, Paolo Grieco, Francesco Petrillo, Anca Hermenean, Francesca Simonelli, Michele D’Amico, Claudio Bucolo, Francesca Lazzara, F
Frontiers in Pharmacology.2024;[Epub] CrossRef - Pharmacological mechanism and clinical study of Qiming granules in treating diabetic retinopathy based on network pharmacology and literature review
Yuxia Huang, Jia Wang, Yu Wang, Wei Kuang, Mengjun Xie, Mei Zhang
Journal of Ethnopharmacology.2023; 302: 115861. CrossRef - Vitreous humor proteome: unraveling the molecular mechanisms underlying proliferative and neovascular vitreoretinal diseases
Fátima Milhano dos Santos, Sergio Ciordia, Joana Mesquita, João Paulo Castro de Sousa, Alberto Paradela, Cândida Teixeira Tomaz, Luís António Paulino Passarinha
Cellular and Molecular Life Sciences.2023;[Epub] CrossRef - Protective Effects of Human Pericyte-like Adipose-Derived Mesenchymal Stem Cells on Human Retinal Endothelial Cells in an In Vitro Model of Diabetic Retinopathy: Evidence for Autologous Cell Therapy
Gabriella Lupo, Aleksandra Agafonova, Alessia Cosentino, Giovanni Giurdanella, Giuliana Mannino, Debora Lo Furno, Ivana Roberta Romano, Rosario Giuffrida, Floriana D’Angeli, Carmelina Daniela Anfuso
International Journal of Molecular Sciences.2023; 24(2): 913. CrossRef - Predictive factors for microvascular recovery after treatments for diabetic retinopathy
Junyeop Lee, Yoon-Jeon Kim, Joo-Yong Lee, Young Hee Yoon, June-Gone Kim
BMC Ophthalmology.2023;[Epub] CrossRef - Pathophysiology and diagnosis of diabetic retinopathy: a narrative review
Mohadese Estaji, Bita Hosseini, Saeed Bozorg-Qomi, Babak Ebrahimi
Journal of Investigative Medicine.2023; 71(3): 265. CrossRef - Proteomics profiling of vitreous humor reveals complement and coagulation components, adhesion factors, and neurodegeneration markers as discriminatory biomarkers of vitreoretinal eye diseases
Fátima M. Santos, Sergio Ciordia, Joana Mesquita, Carla Cruz, João Paulo Castro e Sousa, Luís A. Passarinha, Cândida T. Tomaz, Alberto Paradela
Frontiers in Immunology.2023;[Epub] CrossRef - Hypoxia-induced transcriptional differences in African and Asian versus European diabetic cybrids
Andrew H. Dolinko, Marilyn Chwa, Kevin Schneider, Mithalesh K. Singh, Shari Atilano, Jie Wu, M. Cristina Kenney
Scientific Reports.2023;[Epub] CrossRef - Downregulation of plasma microRNA-29c-3p expression may be a new risk factor for diabetic retinopathy
Bora TORUS, Hakan KORKMAZ, Kuyaş H. OZTURK, Fevziye B. ŞİRİN, Mehmet ARGUN, Sonmez ŞEVİK, Levent TÖK
Minerva Endocrinology.2023;[Epub] CrossRef - Short-Term Outcomes of Intravitreal Faricimab Injection for Diabetic Macular Edema
Sentaro Kusuhara, Maya Kishimoto-Kishi, Wataru Matsumiya, Akiko Miki, Hisanori Imai, Makoto Nakamura
Medicina.2023; 59(4): 665. CrossRef - L-type calcium channel blocker increases VEGF concentrations in retinal cells and human serum
Anmol Kumar, Stefan Mutter, Erika B. Parente, Valma Harjutsalo, Raija Lithovius, Sinnakaruppan Mathavan, Markku Lehto, Timo P. Hiltunen, Kimmo K. Kontula, Per-Henrik Groop, Satyajit Mohapatra
PLOS ONE.2023; 18(4): e0284364. CrossRef - Efficacy and safety of curcumin in diabetic retinopathy: A protocol for systematic review and meta-analysis
Liyuan Wang, Jiayu Xu, Tianyang Yu, Hanli Wang, Xiaojun Cai, He Sun, Godwin Ovenseri-Ogbomo
PLOS ONE.2023; 18(4): e0282866. CrossRef - Highly water-soluble diacetyl chrysin ameliorates diabetes-associated renal fibrosis and retinal microvascular abnormality in db/db mice
Young-Hee Kang, Sin-Hye Park, Young Eun Sim, Moon-Sik Oh, Hong Won Suh, Jae-Yong Lee, Soon Sung Lim
Nutrition Research and Practice.2023; 17(3): 421. CrossRef - Preventive and management approach of triptonide, a diterpenoid compound against streptozotocin-induced diabetic retinopathy in Wistar rat model
Chandramohan Govindasamy, Khalid S. Al-Numair, Jun Li, Weibai Chen, Guoqiang Wu
Arabian Journal of Chemistry.2023; 16(9): 105034. CrossRef - New Insights on Dietary Polyphenols for the Management of Oxidative Stress and Neuroinflammation in Diabetic Retinopathy
Gustavo Bernardes Fanaro, Marcelo Rodrigues Marques, Karin da Costa Calaza, Rafael Brito, André Moreira Pessoni, Henrique Rocha Mendonça, Deborah Emanuelle de Albuquerque Lemos, José Luiz de Brito Alves, Evandro Leite de Souza, Marinaldo Pacífico Cavalcan
Antioxidants.2023; 12(6): 1237. CrossRef - Evaluation of Social Platform-Based Continuity of Care in Improving Cognitive and Prognostic Effects of Young Patients with Diabetic Retinopathy
Guo-lan Cao, Ke-jian Chen
Diabetes, Metabolic Syndrome and Obesity.2023; Volume 16: 1931. CrossRef - Role of vascular endothelial growth factor B in nonalcoholic fatty liver disease and its potential value
Yu-Qi Li, Lei Xin, Yu-Chi Zhao, Shang-Qi Li, Ya-Nuo Li
World Journal of Hepatology.2023; 15(6): 786. CrossRef - The rs1800469 T/T and rs1800470 C/C genotypes of the TGFB1 gene confer protection against diabetic retinopathy in a Southern Brazilian population
Aline Rodrigues Costa, Cristine Dieter, Luís Henrique Canani, Taís Silveira Assmann, Daisy Crispim
Genetics and Molecular Biology.2023;[Epub] CrossRef - Evaluation of Self-Care in Patients with Diabetic Retinopathy
Songül BİLTEKİN, Züleyha KILIÇ, Şefika Dilek GÜVEN
Turkish Journal of Diabetes and Obesity.2023; 7(3): 214. CrossRef - Diabetes mellitus and its influence on the incidence and process of diabetic retinopathy
Janka Poráčová, Melinda Nagy, Marta Mydlárová Blaščáková, Mária Konečná, Vincent Sedlák, Mária Zahatňanská, Tatiana Kimáková, Hedviga Vašková, Viktória Rybárová, Mária Majherová, Ivan Uher
Central European Journal of Public Health.2023; 31(Suppl 1): S4. CrossRef - Dysregulation of the NLRP3 Inflammasome in Diabetic Retinopathy and Potential Therapeutic Targets
Karanvir S. Raman, Joanne A. Matsubara
Ocular Immunology and Inflammation.2022; 30(2): 470. CrossRef - Adult-induced genetic ablation distinguishes PDGFB roles in blood-brain barrier maintenance and development
Elisa Vazquez-Liebanas, Khayrun Nahar, Giacomo Bertuzzi, Annika Keller, Christer Betsholtz, Maarja Andaloussi Mäe
Journal of Cerebral Blood Flow & Metabolism.2022; 42(2): 264. CrossRef - Investigation on the Q-markers of Bushen Huoxue Prescriptions for DR treatment based on chemometric methods and spectrum-effect relationship
Yueting Yu, Ziyu Zhu, Mengjun Xie, Liping Deng, Xuejun Xie, Mei Zhang
Journal of Ethnopharmacology.2022; 285: 114800. CrossRef - Corneal Confocal Microscopy in Type 1 Diabetes Mellitus: A Six-Year Longitudinal Study
Stuti L. Misra, James A. Slater, Charles N. J. McGhee, Monika Pradhan, Geoffrey D. Braatvedt
Translational Vision Science & Technology.2022; 11(1): 17. CrossRef - Microphysiological Neurovascular Barriers to Model the Inner Retinal Microvasculature
Thomas L. Maurissen, Georgios Pavlou, Colette Bichsel, Roberto Villaseñor, Roger D. Kamm, Héloïse Ragelle
Journal of Personalized Medicine.2022; 12(2): 148. CrossRef - Long-Term Oral Administration of Salidroside Alleviates Diabetic Retinopathy in db/db Mice
Fei Yao, Xinyi Jiang, Ling Qiu, Zixuan Peng, Wei Zheng, Lexi Ding, Xiaobo Xia
Frontiers in Endocrinology.2022;[Epub] CrossRef - Classification of macular abnormalities using a lightweight CNN-SVM framework
Xuqian Wang, Yu Gu
Measurement Science and Technology.2022; 33(6): 065702. CrossRef - Chorioretinal Hypoxia Detection Using Lipid-Polymer Hybrid Organic Room-Temperature Phosphorescent Nanoparticles
Yingying Zeng, Van Phuc Nguyen, Yanxiu Li, Do Hyun Kang, Yannis M. Paulus, Jinsang Kim
ACS Applied Materials & Interfaces.2022; 14(16): 18182. CrossRef - LncRNA FLG-AS1 Mitigates Diabetic Retinopathy by Regulating Retinal Epithelial Cell Inflammation, Oxidative Stress, and Apoptosis via miR-380-3p/SOCS6 Axis
Rong Luo, Lan Li, Fan Xiao, Jinsong Fu
Inflammation.2022; 45(5): 1936. CrossRef - Correlation between the progression of diabetic retinopathy and inflammasome biomarkers in vitreous and serum – a systematic review
Charisse Y. J. Kuo, Rinki Murphy, Ilva D. Rupenthal, Odunayo O. Mugisho
BMC Ophthalmology.2022;[Epub] CrossRef - The relationship between the neutrophil-to-lymphocyte ratio and diabetic retinopathy in adults from the United States: results from the National Health and nutrition examination survey
Xiaojie He, Shanshan Qi, Xi Zhang, Jiandong Pan
BMC Ophthalmology.2022;[Epub] CrossRef - Th22 cells induce Müller cell activation via the Act1/TRAF6 pathway in diabetic retinopathy
Yufei Wang, Hongdan Yu, Jing Li, Wenqiang Liu, Shengxue Yu, Pan Lv, Lipan Zhao, Xiaobai Wang, Zhongfu Zuo, Xuezheng Liu
Cell and Tissue Research.2022; 390(3): 367. CrossRef - Prevalence and Factors Associated with Diabetic Retinopathy among Adult Diabetes Patients in Southeast Ethiopia: A Hospital-Based Cross-Sectional Study
Biniyam Sahiledengle, Tesfaye Assefa, Wogene Negash, Anwar Tahir, Tadele Regasa, Yohannes Tekalegn, Ayele Mamo, Zinash Teferu, Damtew Solomon, Habtamu Gezahegn, Kebebe Bekele, Demisu Zenbaba, Alelign Tasew, Fikreab Desta, Zegeye Regassa, Zegeye Feleke, Ch
Clinical Ophthalmology.2022; Volume 16: 3527. CrossRef - Diabetic Retinopathy: Are lncRNAs New Molecular Players and Targets?
Simona Cataldi, Mariagiovanna Tramontano, Valerio Costa, Marianna Aprile, Alfredo Ciccodicola
Antioxidants.2022; 11(10): 2021. CrossRef - Diosgenin protects retinal pigment epithelial cells from inflammatory damage and oxidative stress induced by high glucose by activating AMPK/Nrf2/HO‐1 pathway
Yang Hao, Xuefeng Gao
Immunity, Inflammation and Disease.2022;[Epub] CrossRef - Selective Activation of the Wnt-Signaling Pathway as a Novel Therapy for the Treatment of Diabetic Retinopathy and Other Retinal Vascular Diseases
Huy Nguyen, Sung-Jin Lee, Yang Li
Pharmaceutics.2022; 14(11): 2476. CrossRef - Development and validation of a predictive risk model based on retinal geometry for an early assessment of diabetic retinopathy
Minglan Wang, Xiyuan Zhou, Dan Ning Liu, Jieru Chen, Zheng Zheng, Saiguang Ling
Frontiers in Endocrinology.2022;[Epub] CrossRef - Characterization of NLRP3 Inflammasome Activation in the Onset of Diabetic Retinopathy
Charisse Y-J. Kuo, Jack J. Maran, Emma G. Jamieson, Ilva D. Rupenthal, Rinki Murphy, Odunayo O. Mugisho
International Journal of Molecular Sciences.2022; 23(22): 14471. CrossRef - VEGF Gene Polymorphism Among Diabetes Mellitus and Diabetic Retinopathy
Samra Anees, Saima Shareef, Muhammad Roman, Shah Jahan
Futuristic Biotechnology.2022; : 02. CrossRef - Th22 Cells Induce Müller Cells Activation Via the Act1/Traf6 Pathway in Diabetic Retinopathy
YuFei Wang, Hongdan Yu, Jing Li, Wenqiang Liu, Shengxue Yu, Pan Lv, Lipan Zhao, Xiaobai Wang, Zhongfu Zuo, Xuezheng Liu
SSRN Electronic Journal .2022;[Epub] CrossRef - Pathogenesis of diabetic macular edema: the role of pro-inflammatory and vascular factors. Aliterature review
M.L. Kyryliuk, S.A. Suk
INTERNATIONAL JOURNAL OF ENDOCRINOLOGY (Ukraine).2022; 18(3): 180. CrossRef - Effects of emixustat hydrochloride in patients with proliferative diabetic retinopathy: a randomized, placebo-controlled phase 2 study
Ryo Kubota, Chirag Jhaveri, John M. Koester, Jeffrey K. Gregory
Graefe's Archive for Clinical and Experimental Ophthalmology.2021; 259(2): 369. CrossRef - Transient receptor potential vanilloid 4 channel deletion regulates pathological but not developmental retinal angiogenesis
Holly C. Cappelli, Brianna D. Guarino, Anantha K. Kanugula, Ravi K. Adapala, Vidushani Perera, Matthew A. Smith, Sailaja Paruchuri, Charles K. Thodeti
Journal of Cellular Physiology.2021; 236(5): 3770. CrossRef - Involvement of miR‐126 rs4636297 and miR‐146a rs2910164 polymorphisms in the susceptibility for diabetic retinopathy: a case–control study in a type 1 diabetes population
Eloísa Toscan Massignam, Cristine Dieter, Felipe Mateus Pellenz, Taís Silveira Assmann, Daisy Crispim
Acta Ophthalmologica.2021;[Epub] CrossRef - Retinal Vascular Endothelial Cell Dysfunction and Neuroretinal Degeneration in Diabetic Patients
Malgorzata Mrugacz, Anna Bryl, Katarzyna Zorena
Journal of Clinical Medicine.2021; 10(3): 458. CrossRef - Factors based on optical coherence tomography correlated with vision impairment in diabetic patients
Hiroaki Endo, Satoru Kase, Hikari Tanaka, Mitsuo Takahashi, Satoshi Katsuta, Yasuo Suzuki, Minako Fujii, Susumu Ishida, Manabu Kase
Scientific Reports.2021;[Epub] CrossRef - Class-3 semaphorins: Potent multifunctional modulators for angiogenesis-associated diseases
Bo Jiao, Shiyang Liu, Xi Tan, Pei Lu, Danning Wang, Hui Xu
Biomedicine & Pharmacotherapy.2021; 137: 111329. CrossRef - Single-Cell Analysis of Blood-Brain Barrier Response to Pericyte Loss
Maarja A. Mäe, Liqun He, Sofia Nordling, Elisa Vazquez-Liebanas, Khayrun Nahar, Bongnam Jung, Xidan Li, Bryan C. Tan, Juat Chin Foo, Amaury Cazenave-Gassiot, Markus R. Wenk, Yvette Zarb, Barbara Lavina, Susan E. Quaggin, Marie Jeansson, Chengua Gu, David
Circulation Research.2021;[Epub] CrossRef - VEGFR1 signaling in retinal angiogenesis and microinflammation
Akiyoshi Uemura, Marcus Fruttiger, Patricia A. D'Amore, Sandro De Falco, Antonia M. Joussen, Florian Sennlaub, Lynne R. Brunck, Kristian T. Johnson, George N. Lambrou, Kay D. Rittenhouse, Thomas Langmann
Progress in Retinal and Eye Research.2021; 84: 100954. CrossRef - Changes in Gene Expression Profiling and Phenotype in Aged Multidrug Resistance Protein 4-Deficient Mouse Retinas
Kyung Woo Kim, Sentaro Kusuhara, Atsuko Katsuyama-Yoshikawa, Sho Nobuyoshi, Megumi Kitamura, Sotaro Mori, Noriyuki Sotani, Kaori Ueda, Wataru Matsumiya, Akiko Miki, Takuji Kurimoto, Hisanori Imai, Makoto Nakamura
Antioxidants.2021; 10(3): 455. CrossRef - Circular RNAs: Novel target of diabetic retinopathy
Huan-ran Zhou, Hong-yu Kuang
Reviews in Endocrine and Metabolic Disorders.2021; 22(2): 205. CrossRef - MicroRNA-431-5p encapsulated in serum extracellular vesicles as a biomarker for proliferative diabetic retinopathy
Bo Yu, Mengran Xiao, Fuhua Yang, Jing Xiao, Hui Zhang, Lin Su, Xiaomin Zhang, Xiaorong Li
The International Journal of Biochemistry & Cell Biology.2021; 135: 105975. CrossRef - EndMT Regulation by Small RNAs in Diabetes-Associated Fibrotic Conditions: Potential Link With Oxidative Stress
Roberta Giordo, Yusra M. A. Ahmed, Hilda Allam, Salah Abusnana, Lucia Pappalardo, Gheyath K. Nasrallah, Arduino Aleksander Mangoni, Gianfranco Pintus
Frontiers in Cell and Developmental Biology.2021;[Epub] CrossRef - Pharmacokinetics of genistein distribution in blood and retinas of diabetic and non-diabetic rats
T. Hakami, M.I. Mahmoud, E. de Juan, M. Cooney
Drug Metabolism and Pharmacokinetics.2021; 39: 100404. CrossRef - Nimbolide ameliorates the streptozotocin-induced diabetic retinopathy in rats through the inhibition of TLR4/NF-κB signaling pathway
Xiangwen Shu, Yali Hu, Chao Huang, Ning Wei
Saudi Journal of Biological Sciences.2021; 28(8): 4255. CrossRef - Basic regulatory effects and clinical value of metalloproteinase-14 and extracellular matrix metalloproteinase inducer in diabetic retinopathy
Shuyan Li, Shiheng Lu, Lei Zhang, Shasha Liu, Lei Wang, Kai Lin, Jialun Du, Meixia Song
Materials Express.2021; 11(6): 873. CrossRef - Reduced Acrolein Detoxification in akr1a1a Zebrafish Mutants Causes Impaired Insulin Receptor Signaling and Microvascular Alterations
Haozhe Qi, Felix Schmöhl, Xiaogang Li, Xin Qian, Christoph T. Tabler, Katrin Bennewitz, Carsten Sticht, Jakob Morgenstern, Thomas Fleming, Nadine Volk, Ingrid Hausser, Elena Heidenreich, Rüdiger Hell, Peter Paul Nawroth, Jens Kroll
Advanced Science.2021;[Epub] CrossRef - The Metaflammatory and Immunometabolic Role of Macrophages and Microglia in Diabetic Retinopathy
Honglian Wu, Mengqi Wang, Xiaorong Li, Yan Shao
Human Cell.2021; 34(6): 1617. CrossRef - Inflammatory resolution and vascular barrier restoration after retinal ischemia reperfusion injury
Steven F. Abcouwer, Sumathi Shanmugam, Arivalagan Muthusamy, Cheng-mao Lin, Dejuan Kong, Heather Hager, Xuwen Liu, David A. Antonetti
Journal of Neuroinflammation.2021;[Epub] CrossRef - Diferenças de mensuração de acuidade visual e velocidade de leitura para perto entre pacientes com retinopatia diabética. Repercussão entre conceitos de deficiência visual parcial e cegueira legal
Roberta Freitas Momenté, Isabella Couto Amaral, Luiz Guilherme Coimbra de Brito, João Gabriel Volpato Ferraresi, Maria Luisa Gois da Fonsêca, Nadyr Antônia Damasceno, Luiz Claudio Santos de Souza Lima, Mauricio Bastos Pereira, Eduardo de França Damasceno
Revista Brasileira de Oftalmologia.2021;[Epub] CrossRef - Maintaining blood retinal barrier homeostasis to attenuate retinal ischemia-reperfusion injury by targeting the KEAP1/NRF2/ARE pathway with lycopene
Hao Huang, Xielan Kuang, Xiaobo Zhu, Hao Cheng, Yuxiu Zou, Han Du, Han Tang, Linbin Zhou, Jingshu Zeng, Huijun Liu, Jianhua Yan, Chongde Long, Huangxuan Shen
Cellular Signalling.2021; 88: 110153. CrossRef - Luteolin, an aryl hydrocarbon receptor antagonist, alleviates diabetic retinopathy by regulating the NLRP/NOX4 signalling pathway: Experimental and molecular docking study
Y. Yang, M. Zhou, H. Liu
Physiology International.2021; 108(2): 172. CrossRef - ALDH2/SIRT1 Contributes to Type 1 and Type 2 Diabetes-Induced Retinopathy through Depressing Oxidative Stress
Mengshan He, Pan Long, Tao Chen, Kaifeng Li, Dongyu Wei, Yufei Zhang, Wenjun Wang, Yonghe Hu, Yi Ding, Aidong Wen, Daniela Ribeiro
Oxidative Medicine and Cellular Longevity.2021; 2021: 1. CrossRef - Looking Ahead: Visual and Anatomical Endpoints in Future Trials of Diabetic Macular Ischemia
Chui Ming Gemmy Cheung, Elizabeth Pearce, Beau Fenner, Piyali Sen, Victor Chong, Sobha Sivaprasad
Ophthalmologica.2021; 244(5): 451. CrossRef - The effect of psychotherapy on anxiety, depression, and quality of life in patients with diabetic retinopathy
Suiping Li, Hong Liu, Xian Zhu
Medicine.2021; 100(51): e28386. CrossRef - Changes in Ocular Blood Flow after Ranibizumab Intravitreal Injection for Diabetic Macular Edema Measured Using Laser Speckle Flowgraphy
Lisa Toto, Federica Evangelista, Pasquale Viggiano, Emanuele Erroi, Giada D’Onofrio, Daniele Libertini, Annamaria Porreca, Rossella D’Aloisio, Parravano Mariacristina, Luca Di Antonio, Marta Di Nicola, Rodolfo Mastropasqua
BioMed Research International.2020; 2020: 1. CrossRef - microRNA Expression Profile in the Vitreous of Proliferative Diabetic Retinopathy Patients and Differences from Patients Treated with Anti-VEGF Therapy
Julian Friedrich, David H. W. Steel, Reinier O. Schlingemann, Michael J. Koss, Hans-Peter Hammes, Guido Krenning, Ingeborg Klaassen
Translational Vision Science & Technology.2020; 9(6): 16. CrossRef - Update on the Effects of Antioxidants on Diabetic Retinopathy: In Vitro Experiments, Animal Studies and Clinical Trials
Jose Javier Garcia-Medina, Elena Rubio-Velazquez, Elisa Foulquie-Moreno, Ricardo P Casaroli-Marano, Maria Dolores Pinazo-Duran, Vicente Zanon-Moreno, Monica del-Rio-Vellosillo
Antioxidants.2020; 9(6): 561. CrossRef - Methylglyoxal, a Highly Reactive Dicarbonyl Compound, in Diabetes, Its Vascular Complications, and Other Age-Related Diseases
C. G. Schalkwijk, C. D. A. Stehouwer
Physiological Reviews.2020; 100(1): 407. CrossRef - Pericytes, inflammation, and diabetic retinopathy
Benjamin G. Spencer, Jose J. Estevez, Ebony Liu, Jamie E. Craig, John W. Finnie
Inflammopharmacology.2020; 28(3): 697. CrossRef - Eye hemodynamic data and biochemical parameters of the lacrimal fluid of patients with non-proliferative diabetic retinopathy
Guzal Kangilbaeva, Fazilat Bakhritdinova, Iroda Nabieva, Aziza Jurabekova
Data in Brief.2020; 32: 106237. CrossRef - Endocannabinoids in aqueous humour of patients with or without diabetes
Patrick Richardson, Catherine Ortori, Dave Barrett, Saoirse O'Sullivan, Iskandar Idris
BMJ Open Ophthalmology.2020; 5(1): e000425. CrossRef - A pyruvate dehydrogenase kinase inhibitor prevents retinal cell death and improves energy metabolism in rat retinas after ischemia/reperfusion injury
Kota Sato, Seiya Mochida, Daisuke Tomimoto, Takahiro Konuma, Naoki Kiyota, Satoru Tsuda, Yukihiro Shiga, Kazuko Omodaka, Toru Nakazawa
Experimental Eye Research.2020; 193: 107997. CrossRef - The Role of Bone Morphogenetic Proteins in Diabetic Complications
Nimna Perera, Rebecca H. Ritchie, Mitchel Tate
ACS Pharmacology & Translational Science.2020; 3(1): 11. CrossRef - Increased Ephrin-B2 expression in pericytes contributes to retinal vascular death in rodents
Maha Coucha, Amy C. Barrett, Joseph Bailey, Maryam Abdelghani, Mohammed Abdelsaid
Vascular Pharmacology.2020; 131: 106761. CrossRef - Mitochondrial Defects Drive Degenerative Retinal Diseases
Deborah A. Ferrington, Cody R. Fisher, Renu A. Kowluru
Trends in Molecular Medicine.2020; 26(1): 105. CrossRef - Fli1 deficiency induces endothelial adipsin expression, contributing to the onset of pulmonary arterial hypertension in systemic sclerosis
Takuya Miyagawa, Takashi Taniguchi, Ryosuke Saigusa, Maiko Fukayama, Takehiro Takahashi, Takashi Yamashita, Megumi Hirabayashi, Shunsuke Miura, Kouki Nakamura, Ayumi Yoshizaki, Shinichi Sato, Yoshihide Asano
Rheumatology.2020; 59(8): 2005. CrossRef - Natriuretic Peptides Attenuate Retinal Pathological Neovascularization Via Cyclic Guanosine Monophosphate Signaling in Pericytes and Astrocytes
Katarina Špiranec Spes, Sabrina Hupp, Franziska Werner, Franziska Koch, Katharina Völker, Lisa Krebes, Ulrike Kämmerer, Katrin G. Heinze, Barbara M. Braunger, Michaela Kuhn
Arteriosclerosis, Thrombosis, and Vascular Biology.2020; 40(1): 159. CrossRef - Therapeutic investigation of quercetin nanomedicine in a zebrafish model of diabetic retinopathy
Shuai Wang, Shanshan Du, Wenzhan Wang, Fengyan Zhang
Biomedicine & Pharmacotherapy.2020; 130: 110573. CrossRef - The role of CD44 in pathological angiogenesis
Li Chen, Chenying Fu, Qing Zhang, Chengqi He, Feng Zhang, Quan Wei
The FASEB Journal.2020; 34(10): 13125. CrossRef - Associations between alcohol intake and diabetic retinopathy risk: a systematic review and meta-analysis
Chen Chen, Zhaojun Sun, Weigang Xu, Jun Tan, Dan Li, Yiting Wu, Ting Zheng, Derong Peng
BMC Endocrine Disorders.2020;[Epub] CrossRef - Alpha-Smooth Muscle Actin-Positive Perivascular Cells in Diabetic Retina and Choroid
Soo Jin Kim, Sang A. Kim, Yeong A. Choi, Do Young Park, Junyeop Lee
International Journal of Molecular Sciences.2020; 21(6): 2158. CrossRef - The G‐protein‐coupled chemoattractant receptor Fpr2 exacerbates neuroglial dysfunction and angiogenesis in diabetic retinopathy
Ying Yu, Shengding Xue, Keqiang Chen, Yingying Le, Rongrong Zhu, Shiyi Wang, Shuang Liu, Xinliang Cheng, Huaijin Guan, Ji Ming Wang, Hui Chen
FASEB BioAdvances.2020; 2(10): 613. CrossRef - Dual-Acting Antiangiogenic Gene Therapy Reduces Inflammation and Regresses Neovascularization in Diabetic Mouse Retina
Rute S. Araújo, Diogo B. Bitoque, Gabriela A. Silva
Molecular Therapy - Nucleic Acids.2020; 22: 329. CrossRef - The cells involved in the pathological process of diabetic retinopathy
Songtao Yang, Jiaoyue Zhang, Lulu Chen
Biomedicine & Pharmacotherapy.2020; 132: 110818. CrossRef - The Role of Transforming Growth Factor-Beta in Retinal Ganglion Cells with Hyperglycemia and Oxidative Stress
Hsin-Yi Chen, Yi-Jung Ho, Hsiu-Chuan Chou, En-Chi Liao, Yi-Ting Tsai, Yu-Shan Wei, Li-Hsun Lin, Meng-Wei Lin, Yi-Shiuan Wang, Mei-Lan Ko, Hong-Lin Chan
International Journal of Molecular Sciences.2020; 21(18): 6482. CrossRef - Circadian rhythms in diabetic retinopathy: an overview of pathogenesis and investigational drugs
Ashay D. Bhatwadekar, Varun Rameswara
Expert Opinion on Investigational Drugs.2020; 29(12): 1431. CrossRef - Blood-retinal barrier as a converging pivot in understanding the initiation and development of retinal diseases
Xue Yang, Xiao-Wei Yu, Dan-Dan Zhang, Zhi-Gang Fan
Chinese Medical Journal.2020; 133(21): 2586. CrossRef - Immunosubunit β5i Knockout Suppresses Neovascularization and Restores Autophagy in Retinal Neovascularization by Targeting ATG5 for Degradation
Liyang Ji, Li Li, Ying Zhao, Shengqiang Liu, Jingmin Li, Jinsong Zhang, Qi Zhao, Shuai Wang
Investigative Opthalmology & Visual Science.2020; 61(14): 30. CrossRef - Dipeptidyl Peptidase-4 Inhibitors versus Other Antidiabetic Drugs Added to Metformin Monotherapy in Diabetic Retinopathy Progression: A Real World-Based Cohort Study
Yoo-Ri Chung, Kyoung Hwa Ha, Hyeon Chang Kim, Sang Jun Park, Kihwang Lee, Dae Jung Kim
Diabetes & Metabolism Journal.2019; 43(5): 640. CrossRef - Age-related changes of the human retinal vessels: Possible involvement of lipid peroxidation
Tapas Chandra Nag, Meenakshi Maurya, Tara Sankar Roy
Annals of Anatomy - Anatomischer Anzeiger.2019; 226: 35. CrossRef - Human iPSCs-Derived Endothelial Cells with Mutation in HNF1A as a Model of Maturity-Onset Diabetes of the Young
Kachamakova-Trojanowska, Stepniewski, Dulak
Cells.2019; 8(11): 1440. CrossRef - Diabetic Retinopathy–An Underdiagnosed and Undertreated Inflammatory, Neuro-Vascular Complication of Diabetes
Stephen H. Sinclair, Stanley S. Schwartz
Frontiers in Endocrinology.2019;[Epub] CrossRef - Identification of Diagnostic and Prognostic microRNAs for Recurrent Vitreous Hemorrhage in Patients with Proliferative Diabetic Retinopathy
Parviz Mammadzada, Juliette Bayle, Johann Gudmundsson, Anders Kvanta, Helder André
Journal of Clinical Medicine.2019; 8(12): 2217. CrossRef - RANKL blockade suppresses pathological angiogenesis and vascular leakage in ischemic retinopathy
Sangmi Ock, Soyoung Park, Junyeop Lee, Jaetaek Kim
Biochemical and Biophysical Research Communications.2019; 516(2): 350. CrossRef - EndogenousClostridium perfringensPanophthalmitis with Potential Entry Port from Diverticulitis Exacerbated by Proliferative Diabetic Retinopathy
Vamsee Neerukonda, Anny M. S. Cheng, Swetha Dhanireddy, Samuel Alpert, Han Y. Yin
Case Reports in Ophthalmological Medicine.2019; 2019: 1. CrossRef - Attenuation of Retinal Endothelial Vasodilator Function in a Rat Model of Retinopathy of Prematurity
Ayuki Nakano, Asami Mori, Shiho Arima, Daiki Asano, Akane Morita, Kenji Sakamoto, Tohru Nagamitsu, Tsutomu Nakahara
Current Eye Research.2019; 44(12): 1360. CrossRef - Efficacy of fenofibrate for diabetic retinopathy
Xing-jie Su, Lin Han, Yan-Xiu Qi, Hong-wei Liu
Medicine.2019; 98(14): e14999. CrossRef - Nutraceuticals for the Treatment of Diabetic Retinopathy
Maria Grazia Rossino, Giovanni Casini
Nutrients.2019; 11(4): 771. CrossRef - Efficacy of ranibizumab for the treatment of diabetic retinopathy
Yong-bo Ren, Xing-jie Su, Yan-xiu Qi, He-qun Luan, Qi Sun
Medicine.2019; 98(17): e15409. CrossRef
 PubReader
PubReader Cite
Cite