- Current
- Browse
- Collections
-
For contributors
- For Authors
- Instructions to authors
- Article processing charge
- e-submission
- For Reviewers
- Instructions for reviewers
- How to become a reviewer
- Best reviewers
- For Readers
- Readership
- Subscription
- Permission guidelines
- About
- Editorial policy
Articles
- Page Path
- HOME > Diabetes Metab J > Volume 48(2); 2024 > Article
-
Original ArticleBasic Research Glucolipotoxicity Suppressed Autophagy and Insulin Contents in Human Islets, and Attenuation of PERK Activity Enhanced Them in an ATG7-Dependent Manner
-
Seoil Moon1
 , Ji Yoon Lim1, Mirang Lee2, Youngmin Han2, Hongbeom Kim2, Wooil Kwon2, Jin-Young Jang2, Mi Na Kim1, Kyong Soo Park1, Hye Seung Jung1
, Ji Yoon Lim1, Mirang Lee2, Youngmin Han2, Hongbeom Kim2, Wooil Kwon2, Jin-Young Jang2, Mi Na Kim1, Kyong Soo Park1, Hye Seung Jung1 -
Diabetes & Metabolism Journal 2024;48(2):231-241.
DOI: https://doi.org/10.4093/dmj.2022.0366
Published online: September 6, 2023
- 1,652 Views
- 153 Download
1Department of Internal Medicine, Seoul National University Hospital, Seoul, Korea
2Department of Surgery and Cancer Research Institute, Seoul National University Hospital, Seoul, Korea
-
Corresponding author: Hye Seung Jung Division of Endocrinology and Metabolism, Department of Internal Medicine, Seoul National University Hospital, 101 Daehak-ro, Jongno-gu, Seoul 03080, Korea E-mail: jungjhs@gmail.com
Copyright © 2024 Korean Diabetes Association
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
-
Background
- Administration of pancreatic endoplasmic reticulum kinase inhibitor (PERKi) improved insulin secretion and hyperglycemia in obese diabetic mice. In this study, autophagic balance was studied whether to mediate it.
-
Methods
- Human islets were isolated from living patients without diabetes. PERKi GSK2606414 effects were evaluated in the islets under glucolipotoxicity by palmitate. Islet insulin contents and secretion were measured. Autophagic flux was assessed by microtubule associated protein 1 light chain 3 (LC3) conversion, a red fluorescent protein (RFP)-green fluorescent protein (GFP)- LC3 tandem assay, and P62 levels. For mechanical analyses, autophagy was suppressed using 3-methyladenine in mouse islets. Small interfering RNA for an autophagy-related gene autophagy related 7 (Atg7) was transfected to interfere autophagy.
-
Results
- PERKi administration to mice decreased diabetes-induced P62 levels in the islets. Glucolipotoxicity significantly increased PERK phosphorylation by 70% and decreased insulin contents by 50% in human islets, and addition of PERKi (40 to 80 nM) recovered both. PERKi also enhanced glucose-stimulated insulin secretion (6-fold). PERKi up-regulated LC3 conversion suppressed by glucolipotoxicity, and down-regulated P62 contents without changes in P62 transcription, indicating enhanced autophagic flux. Increased autophagosome-lysosome fusion by PERKi was visualized in mouse islets, where PERKi enhanced Atg7 bound to LC3. Suppression of Atg7 eliminated PERKi-induced insulin contents and secretion.
-
Conclusion
- This study provided functional changes of human islets with regard to autophagy under glucolipotoxicity, and suggested modulation of autophagy as an anti-diabetic mechanism of PERKi.
- Eukaryotic translation initiation factor 2 alpha kinase 3 (EIF2AK3), or pancreatic endoplasmic reticulum kinase (PERK), is a transmembranous protein in the endoplasmic reticulum (ER). Because PERK is known to trigger unfolded protein response (UPR) in response to ER stress [1], and it is critical to β-cell homeostasis, roles of PERK have been investigated extensively in the pancreatic β-cells including in human [2,3].
- To regulate ER stress and UPR in various diseases, specific PERK inhibitors (PERKi), GSK2606414 and GSK2656157 were developed [4]. PERKi treatment to pancreatic β-cells at doses above 1,000 nM acutely abrogated glucose-stimulated insulin secretion (GSIS) and calcium dynamics [5], and deregulated protein synthesis leading to ER stress [6]. in vivo treatment of PERKi in the relevant doses induced islet degeneration and hyperglycemia in mice [7,8].
- In contrast to the complete inhibition of PERK activity, we found that partial attenuation of PERK with low doses of PERKi (around 40 nM) rather improved GSIS in pancreatic islets isolated from mice and humans, through binding immunoglobulin protein (BIP)-dependent calcium regulation [9]. In addition, low-dose PERKi had anti-diabetic effects in a mouse model of type 2 diabetes mellitus, induced by a high-fat diet and streptozotocin: oral administration of PERKi significantly improved GSIS and hyperglycemia throughout 8 weeks in the model, which were comparable to effects of sulfonylurea administration [10]. However, underlying mechanisms have not been identified. The effective doses of PERKi seemed somewhat low to inhibit phosphorylation of EIF2A, the downstream to UPR signaling [6,9].
- Macroautophagy (herein, autophagy) is another cellular process playing an essential role in β-cell homeostasis [11]. Dysregulated autophagy has been implicated in diabetes pathophysiology, and its modulation was suggested to have a therapeutic potential in β-cell failure [12,13]. Therefore, we studied whether low-dose PERKi modulates autophagic activity in human islets under glucolipotoxicity (GLT) and whether it is linked to β-cell function.
INTRODUCTION
- Human islet isolation
- We obtained pancreatic tissues from non-diabetic patients who underwent pancreatic surgery for tumors [14]. The study was approved by the Institutional Review Board of Seoul National University Hospital (IRB No. 0901-010-267 and 2110-151-1266). All eight participants (three men, five women) provided written informed consent. The mean±standard deviation of age was 54.5±16.0 years, body mass index 25.6±3.6 kg/m2, and glycosylated hemoglobin 5.7%±0.3% (Supplementary Table 1).
- We isolated islets from 0.80 g (median range, 0.21 to 5.0) of pancreatic tissues that were grossly normal. In less than 1.5 hours stored in histidine-tryptophan-ketoglutarate solution, the tissues were chopped and digested with collagenase NB and neutral protease (SERVA Electrophoresis GmbH, Heidelberg, Germany). Subsequently islets were separated using Ficoll gradients (P04-69600, Pan-biotech, Aidenbach, Germany). After washing the islets with Hanks’ balanced salt solution (HBSS; LB 003-02, WELGENE, Gyeongsan, Korea) containing 10% fetal bovine serum (FBS; 16000-044, ThermoFisher, Waltham, MA, USA), we hand-picked islets and incubated overnight before experiments, as described before [9]. The isolation yield was about 700 islets/g pancreas (Supplementary Table 1).
- Mouse islet isolation
- Islets were isolated from adult C57BL/6J mice via intraductal collagenase digestion technique, hand-picked, and cultured at least 3 hours before experiments [10]. All animal experiments were conducted in accordance with the Institutional Animal Care and Use Committee (SNU-198989-1 and 19-0190-S1A0).
- Culture medium
- Pancreatic islets and dispersed islet cells, from either mice or humans, were incubated in RPMI1640 (LM 011-01, WELGENE) with 10% FBS and penicillin-streptomycin solution (SV30010, HyClone, Cytiva, Marlborough, MA, USA). Final glucose concentration of the culture condition was 10 mM.
- Reagents
- GSK2606414 (#516535, CALBIOCHEM, San Diego, CA, USA) and 3-methyladenine (3-MA; M9281, Sigma, St. Louis, MO, USA) were dissolved in dimethyl sulfoxide (DMSO). Treatment concentrations were determined according to the previous works [9].
- Glucolipotoxicity in vitro
- Intact islets were incubated in the islet culture media, containing 20-mM glucose and 0.5-mM palmitate. Palmitate was conjugated with bovine serum albumin (BSA) free from fatty acid (3:1 molar ratio) (Sigma-Aldrich).
- GSIS and insulin content
- Human islets (with diameter 100 to 150 μm) or mouse islet cells (1×105 cells/well) were used for the experiments. They were fasted for 40 minutes in KRB HEPES (KRBH) buffer containing 0.2% BSA and low glucose (LG, 1.67 mM for human islets; 2.8 mM for mouse islet cells) at 37°C. Then the media were replaced with fresh LG-KRBH, incubation for 40 minutes, collection of the supernatants, and replaced again with high glucose (HG, 17.5 mM) KRBH. After 40 minutes of further incubation, we collected the supernatants and assayed insulin using an enzyme-linked immunosorbent assay (ELISA) kit (80-INSHU-E01.1 for human insulin; 80-INSMS-E01 for mouse insulin, ALPCO, Salem, NH, USA). After the supernatants were collected, the cells were collected, dissolved in 0.2 M HCl in 75% ethanol, and sonicated. We then neutralized the lysates with 0.2 M NaOH, collected the supernatants, and assayed insulin using the same ELISA kits.
- Western blotting
- We extracted total protein from homogenized islets using radioimmunoprecipitation assay (RIPA) buffer mixed with protease inhibitor cocktail and phosphatase inhibitor (04 906 845 001, 11697498001, Roche, Basel, Switzerland). We boiled the protein samples in sodium dodecyl sulfate (SDS) sample buffer, separated them by SDS-polyacrylamide gel electrophoresis, and then transferred them to nitrocellulose membranes for immunoblotting. For the microtubule associated protein 1 light chain 3B (LC3B) blots bound to autophagy related 7 (ATG7), non-reducing gel was used. We detected the blots using the ECL Western blotting substrate (RPN2232, Amersham, Stafford, UK). Primary antibodies used are presented in the Supplementary Table 2.
- Quantitative reverse transcription-polymerase chain reaction
- We extracted total RNA using an extraction kit (Intronbio, Seongnam, Korea), performed reverse transcription using GoScript system (A5001, Promega, Madison, WI, USA), and quantitative polymerase chain reaction (PCR) using GoTaq qPCR Master Mix (A6001, Promega) and the Applied Biosystems 7500 real-time PCR Instrument system (Waltham, MA, USA). Primer sequences are presented in Supplementary Table 3. We normalized the data with the mRNA of 18s.
- RNA interfering
- We dissociated mouse islets into single cells using trypsin (ThermoFisher Scientific Inc.) and transfected the cells with 100 nM of Atg7 siRNA (siAtg7; 4390816, Ambion, Foster City, CA, USA) or of negative control siRNA (siNS; SN-1003, Bioneer, Daejeon, Korea) using Lipofectamine RNAiMAX (13778-150, Invitrogen, Waltham, MA, USA) according to the manufacturer’s instructions. After 2 to 3 days, RNA and proteins were extracted from the cells.
- P62 staining intensity
- Paraffin blocks embedding pancreas from the previous study [10] were cut and placed on slides. The slides were stained using the Discovery XT automated immunohistochemistry stainer (Ventana Medical Systems Inc., Tucson, AZ, USA). Detection was done using the Ventana ChromoMap Kit (Ventana Medical Systems). The stained sections were analyzed using Aperio Digital Pathology and ImageScope (Leica Biosystems, Wetzlar, Germany). About 10.1±1.8 islets were counted in every mouse for the measurement of P62 statin intensity, and quantification of the intensity per islet area was calculated using ImageJ (National Institutes of Health, Bethesda, MD, USA).
- Tandem fluorescent RFP-GFP-LC3 assay
- Mouse islet cells were dispersed, transfected with Premo autophagy tandem sensor red fluorescent protein (RFP)-green fluorescent protein (GFP)-LC3B kit (Cat. P36239, Invitrogen) and plated on a chambered slide glass. After overnight incubation, the cells were treated with 0.5-mM 3-MA with and without 20-nM PERKi for 29 hours. During the last 5 hours, serum was deprived to induce autophagy. Before fixation with 4% formaldehyde, live cell nuclei were stained with Hoechst 33342, and the cells were visualized by a confocal laser scanning fluorescent microscope (DMi8, inverted) using a Leica TCS SP8 system. Cytoplasmic GFP and RFP puncta were manually counted among live cells with distinct nuclei, to exclude cell death-associated autophagy.
- Statistics
- Data are expressed as the mean±standard error of the mean and median (interquartile range). Statistical analyses were executed using Prism 5 software (GraphPad, San Diego, CA, USA), as described in figure legends. P values less than 0.05 were considered statistically significant.
METHODS
- Low-dose PERKi treatment decreased P62 levels in the islets of obese diabetic mice
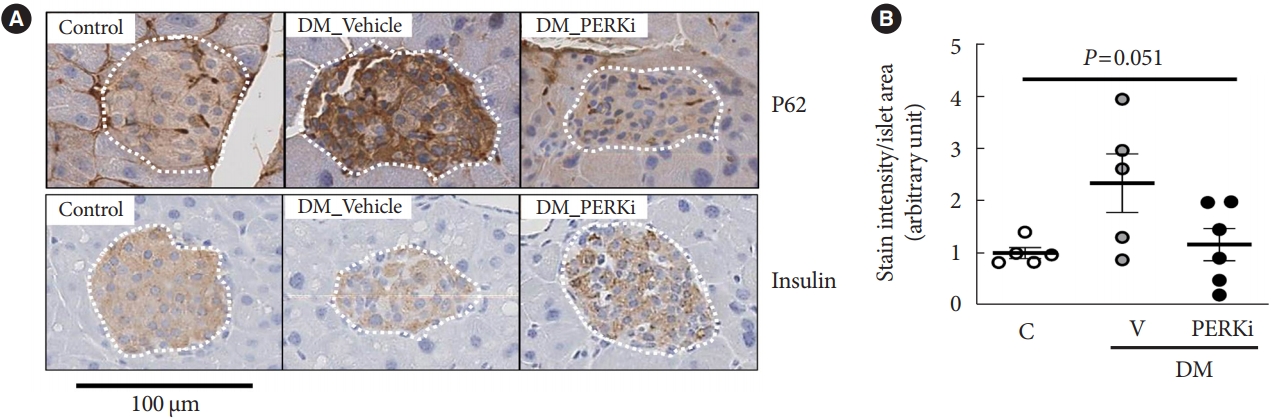
- First, we tried immunohistochemical stain for P62 which is removed by autophagy [15], in the pancreas tissues of the mice mimicking type 2 diabetes mellitus mentioned in the introduction (blood glucose and insulin levels in Supplementary Table 4) [10]. The stain intensity in the cytoplasm of islet cells was higher in the diabetic mice compared to non-diabetic control mice, and PERKi treatment to diabetic mice for 8 weeks seemed to reduce the intensity (Fig. 1), suggesting that in vivo PERKi might have enhanced autophagic flux in the islets. Therefore, further experiments were performed in vitro. Insulin staining intensity (Fig. 1), β-cell mass, and pancreatic insulin contents [10] were not affected significantly by chronic administration of PERKi.
- Low-dose PERKi enhanced autophagic flux in human islets under GLT
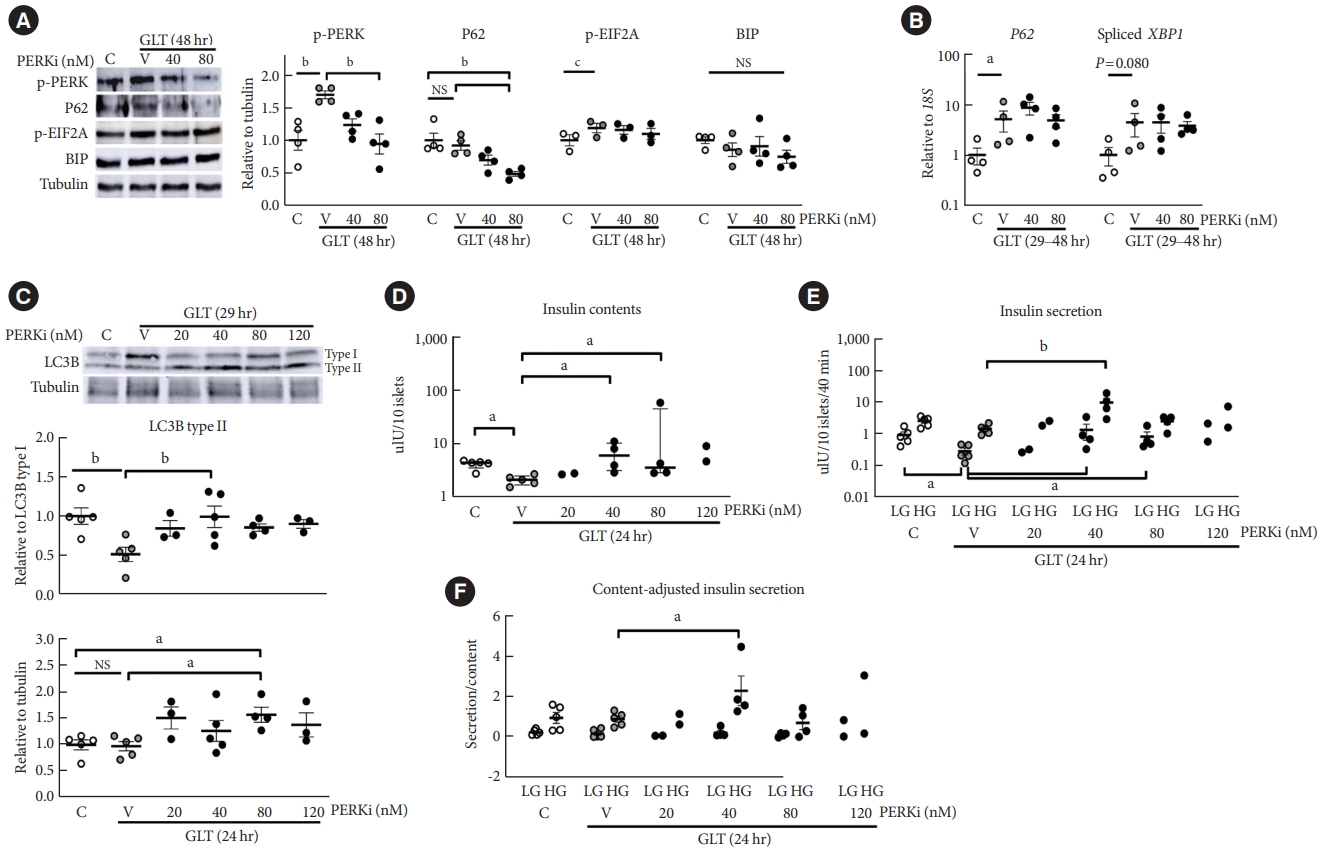
- GLT increased PERK phosphorylation, and addition of lowdose PERKi reduced it dose-dependently (P<0.01) (Fig. 2A). Even though GLT for 48 hours did not increase P62 compared to the control, PERKi induced step-wise reductions in P62 levels to reach statistical significance at 80 nM (P<0.01), compatible with the in vivo PERKi (Fig. 1). EIF2A was slightly activated by GLT, but not influenced by PERKi significantly. BIP amount did not change by either (Fig. 2A, Supplementary Fig. 1). According to quantitative reverse transcription-polymerase chain reaction (RT-PCR), GLT increased P62 transcript and spliced mRNA of X-box binding protein 1 (XBP1); however, addition of PERKi did not alter the levels (Fig. 2B). Collectively, GLT triggered PERK-EIF2A pathway along with spliced-XBP1 and P62 transcription. Treatment of PERKi as low as 80 nM reduced the PERK activation but not the EIF2A activity or XBP1 splicing. GLT-induced P62 expression was not influenced by PERKi; therefore, decline of P62 protein levels by PERKi suggests enhanced clearance.
- Next, LC3 lipidation, a necessary process of autophagosome formation, was examined after shorter exposure to GLT to observe antecedent events to the P62 reduction. As for effects of GLT on LC3B lipidation, ratio to LC3B type I was suppressed (P<0.01) while the amount relative to tubulin was not affected (Fig. 2B, upper and lower panels). However, LC3 lipidation was significantly enhanced by 40 to 80-nM PERKi, estimated by either way (P<0.05) (Fig. 2C). These results suggested that exposure to GLT induced ER stress, triggering PERK phosphorylation in human islets, and attenuation of it using low-dose PERKi enhanced autophagic flux.
- Low-dose PERKi improved human islet function under GLT
- Exposure to GLT for 24 hours significantly decreased insulin contents, which were increased by PERKi at 40 to 80 nM (all P<0.05) (Fig. 2D). The changes in insulin contents were mirrored by those in the ratio of LC3B-II to I (Fig. 2C, upper panel), and those in insulin secretion during low-glucose condition (Fig. 2E). Prolonged exposure to GLT could have rather increased basal insulin secretion, as Marselli et al. [16] reported.
- Indeed, GLT significantly inhibited GSIS (Supplementary Fig. 2: the same data in Fig. 2D-F). However, comparing with the prominent effects by PERKi influenced the statistical results, masking the significant impairment of GSIS by GLT (Fig. 2E). Addition of 40-nM PERKi not only recovered GLT-impaired GSIS but also overshoot the control in some cases. Adjusting the secretion by insulin contents maintained the augmented GSIS (Fig. 2F).
- Low-dose PERKi recovered insulin synthesis through ATG7
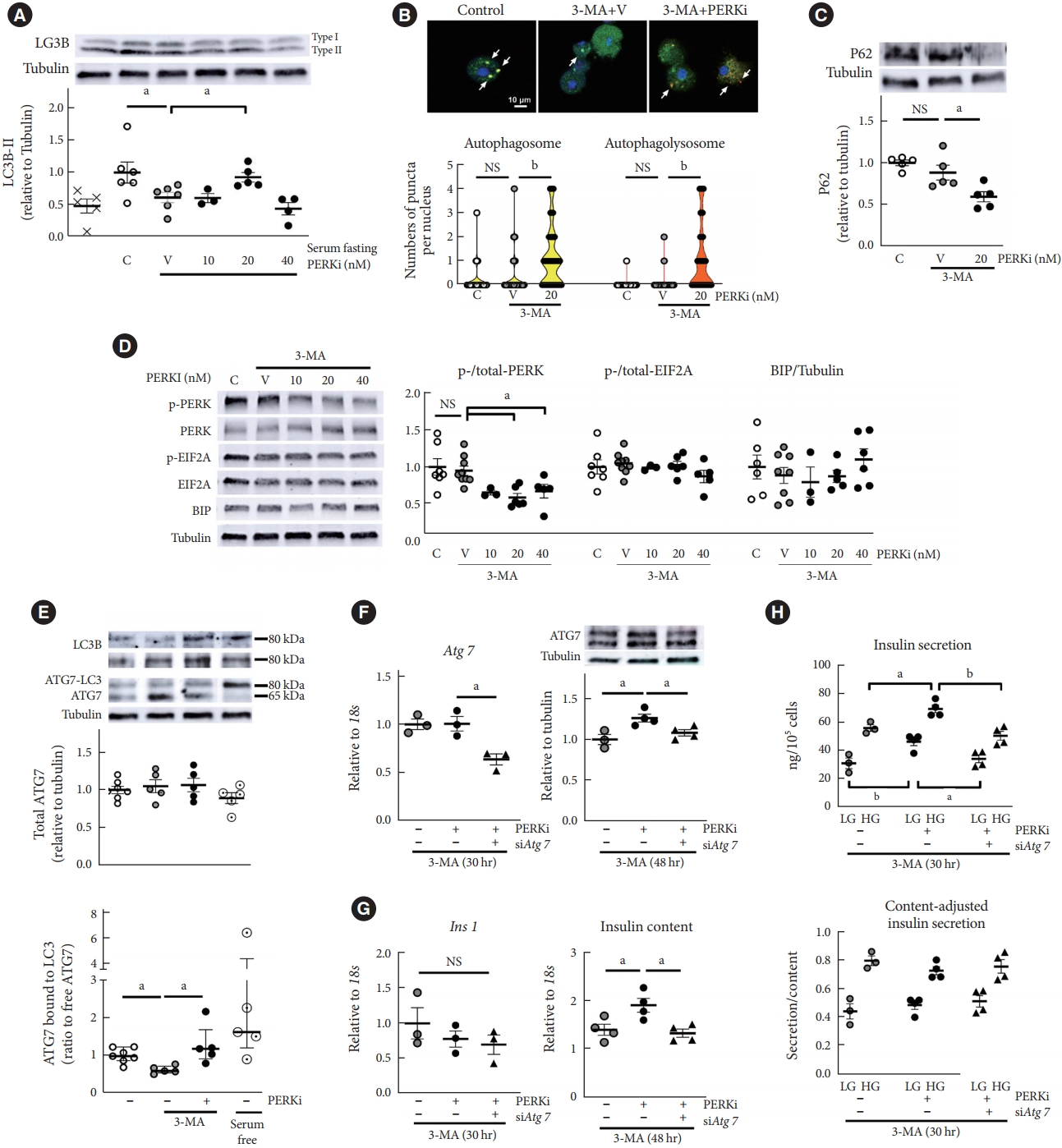
- GLT can stimulate various pathways besides autophagy, including ER stress (Fig. 2A and B) [17], then PERKi effect was examined in the condition where autophagy was specifically inhibited by 3-MA. 3-MA is a phosphatidylinositol 3 (PI3) kinase inhibitor and interferes initiation of autophagosome formation. Although impaired autophagy can induce ER stress theoretically [3], chronic treatment of low-dose 3-MA has not been demonstrated to induce ER stress. In the context of starvation-induced autophagy in mouse islets, 3-MA (0.5 mM) treatment significantly reduced LC3B type II relative to tubulin (Fig. 3A). However, it did not either decrease numbers of autophagolysosome (Fig. 3B) or induce P62 accumulation (Fig. 3C). chronic treatment period might have activated compensatory mechanisms such as enhanced LC3 production, reflected by dispersed GFP signals in the cytoplasm (Fig. 3B). PERK phosphorylation was not stimulated either (Fig. 3D), and in this system where UPR was not triggered, attenuation of PERK activity by low-dose PERKi replicated induction of autophagic flux (P<0.05) (Fig. 3A-C). Compared to the human islets, mouse islets showed the responses at lower concentrations of PERKi, as was observed [9].
- From these findings, PERKi was presumed to act in the formation of autophagosome, possibly through LC3 lipidation [18]. Then, PERKi influences on Atg7 was investigated, as it is a critical enzyme for LC3 lipidation (Supplementary Fig. 3) [11,19]. PERKi enhanced not total Atg7 amount but ratio of ATG7-LC3 complex to free ATG7, which was interfered by 3-MA (Fig. 3E). Representative blots under non-reducing condition are presented: high molecular-weight Atg7 bound to LC3 (approximately an additional 15 kDa), along with LC3B blots on two different gels [20]. To understand its relevance to autophagic activity, serum was deprived for 5 hours as a positive control of autophagic activation, and it increased proportion of ATG7-LC3 complex (Fig. 3E, lower panel). When Atg7 expression was down-regulated using siRNA (Fig. 3F), insulin 1 (Ins1) mRNA levels did not change, but PERKi-induced insulin contents and insulin secretion were abrogated (Fig. 3G and H). It suggested that PERKi-induced insulin contents through Atg7 did not result from promotion of proinsulin transcription, but of translation/post-translational modification.
- Collectively, we speculated that PERKi-induced regulation of autophagy would be mediated by modulation of thioester bindings between Atg7 and LC3 [18].
RESULTS
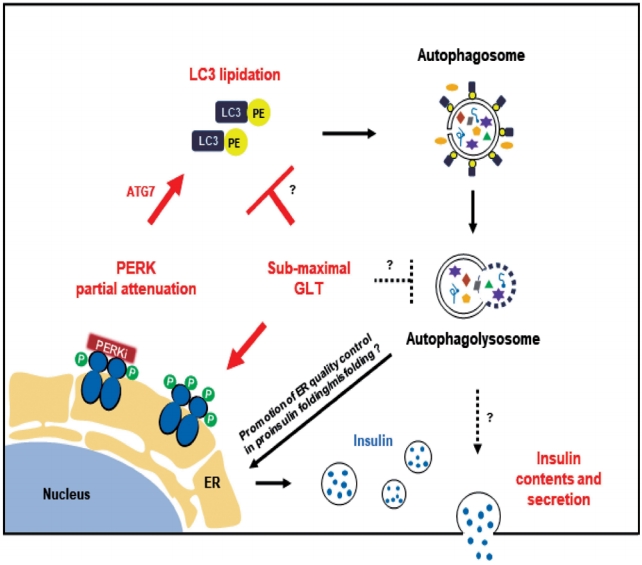
- In this study using human islets, we observed that GLT for 24 hours reduced insulin contents and insulin secretion, and low-dose PERKi recovered them (Fig. 2D-F, Supplementary Fig. 2), which had been observed in islets without metabolic stress, too [9]. Changes in the β-cell function by PERKi under GLT were correlated with those in autophagic flux, including LC3B conversion (Fig. 2A-C). Because PERKi-induced β-cell function was dependent on Atg7 (Fig. 3G and H), and Atg7 has a key role in LC3 lipidation and biogenesis of autophagosomes [18], we propose that PERKi-induced autophagy contributed to the PERKi-recovered β-cell function under GLT.
- Strength of this study is the cells we used, which are human islets from living donors (Supplementary Table 1) [21] and would have a merit with respect to clinical translation of the results. Fresh pancreatic tissues from living donors would represent physiologic islet cells better than those from deceased organ donors, through minimizing the metabolic changes associated with brain death and the autolysis of pancreatic tissues after death [22]. The participants in this study underwent diagnostic or preemptive pancreatectomy for pre-malignant diseases, so none had been prescribed with anti-cancer treatment, glucocorticoids, or vasopressors before the surgery. In addition, the pancreas samples were exposed to very short duration of cold ischemia, and it might be especially critical to describe autophagic process which is highly susceptible to cellular homeostasis and viability [19,23]. We used pure islets by hand-picking, then the study results would be free from confounding effects of digested exocrine tissues. Weaknesses of the study is insufficiency of mechanistic analyses using the human cells from limited supply, then we performed it using mouse islets.
- Our observations were different from other groups’ using human islets from deceased organ donors, where LC3 lipidation, density volume of autophagosomes, P62 levels, and cell death had been increased by GLT [24-28]. This phenomenon was explained by insufficient lysosomal function due to decreased acidity [29]; that is, primary insult by GLT would be autophagolysosomal degradation, not autophagosome formation. However, LC3B lipidation seemed suppressed by GLT in the current study (Fig. 2C). Considering that P62 levels were not raised by GLT compared to the control even though P62 mRNA was induced (Fig. 2A and B), GLT inhibited LC3 conversion but not deteriorate autophagic flux significantly. Several reasons can be assumed as follows: adaptive UPR to GLT, reflected by enhanced EIF2A phosphorylation and spliced-XBP1 (Fig. 2A and B), might have precluded P62 translation; the GLT condition was less toxic (20-mM glucose, 0.5-mM palmiate, 29 to 48 hours) than those in other groups (higher concentrations and/or longer duration), while the control condition was higher in the glucose levels (10 mM) than other groups (usually 5.5 mM), where P62 levels could have already increased in human islets [28]; finally, islets from living donors might be more resistant to GLT than those from deceased ones as mentioned in the previous paragraph, even though we cannot prove it here because use of islets from organ donors for research purpose is prohibited in Korea. Interestingly, palmitate exposure for just 14 hours significantly accumulate P62 and subsequent LC3 lipidation in INS1 cell lines [29,30], while GLT for 29 hours tended to increase P62 with abrogation of LC3 lipidation in mouse islets Supplementary Fig. 4). LC3 lipidation rebounded after extended GLT for 48 hours [31]. Lipotoxicity with/without HG seems to stimulate variable autophagic responses depending on the cell conditions and sources.
- Human β-cell viability with regard to autophagy under GLT has been repeatedly examined where LC3 lipidation was induced by GLT [24-28]; however, human β-cell function has not been well described with autophagic process. Functional impairment would precede cell death. Even though the GLT in our study may not be toxic enough to induce lysosomal dysfunction and cell death, it was enough to suppress β-cell function and LC3 type II ratio to I, then we could show that PERKi recovered both (Fig. 2). Even though deteriorated GSIS in response to lipotoxicity is generally accepted [17], Roomp et al. [32] reported that exposure to palmitate as long as 7 days did not suppress GSIS in human islets, so in vivo impact of GLT including unsaturated fatty acids on human insulin secretion would be determined deliberately [33].
- In this study, we propose that PERKi-induced autophagic activity would enhance human β-cell function under metabolic stress (Fig. 4). There are many links yet to reveal. First, the link between PERKi and autophagic activity. PERKi-induced ATG7-LC3 thioester bindings (Fig. 3E) might have been mediated by PERK-nuclear factor erythroid-2-related factor 2 (NRF2) pathway which contributes to β-cell redox homeostasis [20,34]. Another explanation could be activating transcription factor 6 (ATF6) branch among the ER stress pathways. Even when ATF6 was not activated, PERKi seemed to reduce cleaved ATF6α, suggesting down-regulation of the pathway (Supplementary Fig. 5), which might be able to attenuate mechanistic target of rapamycin complex 1 (mTORC1) signals leading to activation of autophagic process [35,36]. These are currently under investigation.
- Another question is how the PERKi-induced autophagic activity enhanced insulin contents. Generation and maintenance of the insulin granule pool is regarded predominately regulated at the translational level. Enhanced ER macroautophagy, that is ER-phagy would improve ER turnover under metabolic stress, contributing to the ER quality control in proinsulin folding/misfolding. Especially, ER-phagy is likely to be beneficial in clearing misfolded proinsulin and thereby enhancing efficiency of the remaining proinsulin to be properly folded and to undergo successful intracellular transport. However, hyperactivation of autophagy in a different context can stimulate turnover of mature insulin secretory granules, causing a diminution of the insulin storage pool [37-39].
- Indeed, we cannot totally exclude PERKi-induced de-repression of general translation, even though changes in the phosphorylation of EIF2A was not detected in this experiment setting (Figs. 2A and 3D), warranting confirmatory studies using sophisticated tools. Furthermore, we had demonstrated glucose-lowering effects of PERKi in β-cell specific Atg7-deleted mice [9], which suggests PERKi-triggered signaling other than ATG7-dependent one. Mechanisms of augmented GSIS by PERKi is still unanswered, and future studies on these questions would reinforce potential clinical application of PERKi in diabetes.
DISCUSSION
SUPPLEMENTARY MATERIALS
Supplementary Table 1.
Supplementary Table 2.
Supplementary Table 3.
Supplementary Table 4.
Supplementary Fig. 1.
Supplementary Fig. 2.
Supplementary Fig. 3.
Supplementary Fig. 4.
Supplementary Fig. 5.
-
CONFLICTS OF INTEREST
Kyong Soo Park has been honorary editors of the Diabetes & Metabolism Journal since 2020. He was not involved in the review process of this article. Otherwise, there was no conflict of interest.
-
AUTHOR CONTRIBUTIONS
Conception or design: H.S.J.
Acquisition, analysis, or interpretation of data: S.M., J.Y.L., M.L., Y.H., H.K., W.K., J.Y.J., M.N.K.
Drafting the work or revising: S.M., K.S.P.
Final approval of the manuscript: S.M., J.Y.L., M.L., Y.H., H.K., W.K., J.Y.J., M.N.K., K.S.P., H.S.J.
-
FUNDING
This study was supported by NRF (grant numbers 2019R1A2 C1007397 and 2022R1A2C2004570) funded by the Ministry of Science and ICT, Republic of Korea.
NOTES
-
Acknowledgements
- We thank Prof. Young Joo Park (Seoul National University, Korea) for animal research facility.
- 1. Kaufman RJ. Orchestrating the unfolded protein response in health and disease. J Clin Invest 2002;110:1389-98.ArticlePubMedPMC
- 2. Kefalas G, Larose L. PERK leads a hub dictating pancreatic b cell homoeostasis. Biol Cell 2018;110:27-32.PubMedPDF
- 3. Moon S, Jung HS. Endoplasmic reticulum stress and dysregulated autophagy in human pancreatic beta cells. Diabetes Metab J 2022;46:533-42.ArticlePubMedPMCPDF
- 4. Axten JM, Medina JR, Feng Y, Shu A, Romeril SP, Grant SW, et al. Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl)phenyl] acetyl}-2,3-dihydro-1H-indol-5-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK). J Med Chem 2012;55:7193-207.ArticlePubMed
- 5. Wang R, McGrath BC, Kopp RF, Roe MW, Tang X, Chen G, et al. Insulin secretion and Ca2+ dynamics in b-cells are regulated by PERK (EIF2AK3) in concert with calcineurin. J Biol Chem 2013;288:33824-36.PubMedPMC
- 6. Harding HP, Zyryanova AF, Ron D. Uncoupling proteostasis and development in vitro with a small molecule inhibitor of the pancreatic endoplasmic reticulum kinase, PERK. J Biol Chem 2012;287:44338-44.ArticlePubMedPMC
- 7. Atkins C, Liu Q, Minthorn E, Zhang SY, Figueroa DJ, Moss K, et al. Characterization of a novel PERK kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res 2013;73:1993-2002.ArticlePubMedPDF
- 8. Moreno JA, Halliday M, Molloy C, Radford H, Verity N, Axten JM, et al. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci Transl Med 2013;5:206ra138.ArticlePubMed
- 9. Kim MJ, Min SH, Shin SY, Kim MN, Lee H, Jang JY, et al. Attenuation of PERK enhances glucose-stimulated insulin secretion in islets. J Endocrinol 2018;236:125-36.ArticlePubMed
- 10. Kim MJ, Kim MN, Min SH, Ham DS, Kim JW, Yoon KH, et al. Specific PERK inhibitors enhanced glucose-stimulated insulin secretion in a mouse model of type 2 diabetes. Metabolism 2019;97:87-91.ArticlePubMed
- 11. Jung HS, Chung KW, Kim JW, Kim J, Komatsu M, Tanaka K, et al. Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metab 2008;8:318-24.PubMed
- 12. Kim J, Park K, Kim MJ, Lim H, Kim KH, Kim SW, et al. An autophagy enhancer ameliorates diabetes of human IAPP-transgenic mice through clearance of amyloidogenic oligomer. Nat Commun 2021;12:183.ArticlePubMedPMCPDF
- 13. Marasco MR, Linnemann AK. B-cell autophagy in diabetes pathogenesis. Endocrinology 2018;159:2127-41.PubMedPMC
- 14. Yoon JW, Jung HS, Jang JY, Kim MJ, Kim JH, Ohn JH, et al. Improved insulin secretion by autologous islet transplantation, compared to oral antidiabetic agents, after distal pancreatectomy. Cell Transplant 2015;24:1615-26.ArticlePubMedPDF
- 15. Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016;12:1-222.PubMedPMC
- 16. Marselli L, Piron A, Suleiman M, Colli ML, Yi X, Khamis A, et al. Persistent or transient human b cell dysfunction induced by metabolic stress: specific signatures and shared gene expression with type 2 diabetes. Cell Rep 2020;33:108466.PubMed
- 17. Lytrivi M, Castell AL, Poitout V, Cnop M. Recent insights into mechanisms of b-cell lipo- and glucolipotoxicity in type 2 diabetes. J Mol Biol 2020;432:1514-34.PubMed
- 18. Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, Ishihara N, et al. A ubiquitin-like system mediates protein lipidation. Nature 2000;408:488-92.ArticlePubMedPDF
- 19. Lee YH, Kim J, Park K, Lee MS. B-cell autophagy: mechanism and role in b-cell dysfunction. Mol Metab 2019;27S(Suppl):S92-103.PubMed
- 20. Frudd K, Burgoyne T, Burgoyne JR. Oxidation of Atg3 and Atg7 mediates inhibition of autophagy. Nat Commun 2018;9:95.ArticlePubMedPMCPDF
- 21. Hart NJ, Powers AC. Use of human islets to understand islet biology and diabetes: progress, challenges and suggestions. Diabetologia 2019;62:212-22.ArticlePubMedPMCPDF
- 22. Barovic M, Distler M, Schoniger E, Radisch N, Aust D, Weitz J, et al. Metabolically phenotyped pancreatectomized patients as living donors for the study of islets in health and diabetes. Mol Metab 2019;27S(Suppl):S1-6.ArticlePubMedPMC
- 23. Vivot K, Pasquier A, Goginashvili A, Ricci R. Breaking bad and breaking good: b-cell autophagy pathways in diabetes. J Mol Biol 2020;432:1494-513.PubMed
- 24. Cnop M, Abdulkarim B, Bottu G, Cunha DA, Igoillo-Esteve M, Masini M, et al. RNA sequencing identifies dysregulation of the human pancreatic islet transcriptome by the saturated fatty acid palmitate. Diabetes 2014;63:1978-93.ArticlePubMedPDF
- 25. Masini M, Bugliani M, Lupi R, del Guerra S, Boggi U, Filipponi F, et al. Autophagy in human type 2 diabetes pancreatic beta cells. Diabetologia 2009;52:1083-6.ArticlePubMedPDF
- 26. Martino L, Masini M, Novelli M, Beffy P, Bugliani M, Marselli L, et al. Palmitate activates autophagy in INS-1E b-cells and in isolated rat and human pancreatic islets. PLoS One 2012;7:e36188.ArticlePubMedPMC
- 27. Zummo FP, Krishnanda SI, Georgiou M, O’Harte FP, Parthsarathy V, Cullen KS, et al. Exendin-4 stimulates autophagy in pancreatic b-cells via the RAPGEF/EPAC-Ca2+-PPP3/calcineurin-TFEB axis. Autophagy 2022;18:799-815.ArticlePubMedPMC
- 28. Mir SU, George NM, Zahoor L, Harms R, Guinn Z, Sarvetnick NE. Inhibition of autophagic turnover in b-cells by fatty acids and glucose leads to apoptotic cell death. J Biol Chem 2015;290:6071-85.PubMed
- 29. Trudeau KM, Colby AH, Zeng J, Las G, Feng JH, Grinstaff MW, et al. Lysosome acidification by photoactivated nanoparticles restores autophagy under lipotoxicity. J Cell Biol 2016;214:25-34.ArticlePubMedPMCPDF
- 30. Las G, Serada SB, Wikstrom JD, Twig G, Shirihai OS. Fatty acids suppress autophagic turnover in b-cells. J Biol Chem 2011;286:42534-44.PubMedPMC
- 31. Zummo FP, Cullen KS, Honkanen-Scott M, Shaw JA, Lovat PE, Arden C. Glucagon-like peptide 1 protects pancreatic bcells from death by increasing autophagic flux and restoring lysosomal function. Diabetes 2017;66:1272-85.PubMed
- 32. Roomp K, Kristinsson H, Schvartz D, Ubhayasekera K, Sargsyan E, Manukyan L, et al. Combined lipidomic and proteomic analysis of isolated human islets exposed to palmitate reveals time-dependent changes in insulin secretion and lipid metabolism. PLoS One 2017;12:e0176391.ArticlePubMedPMC
- 33. Weir GC. Glucolipotoxicity, b-cells, and diabetes: the emperor has no clothes. Diabetes 2020;69:273-8.PubMedPMC
- 34. Yagishita Y, Fukutomi T, Sugawara A, Kawamura H, Takahashi T, Pi J, et al. Nrf2 protects pancreatic b-cells from oxidative and nitrosative stress in diabetic model mice. Diabetes 2014;63:605-18.PubMed
- 35. Allen D, Seo J. ER stress activates the TOR pathway through Atf6. J Mol Signal 2018;13:1.ArticlePubMedPMC
- 36. Blackwood EA, Hofmann C, Santo Domingo M, Bilal AS, Sarakki A, Stauffer W, et al. ATF6 regulates cardiac hypertrophy by transcriptional induction of the mTORC1 activator, Rheb. Circ Res 2019;124:79-93.ArticlePubMedPMC
- 37. Levine B, Kroemer G. Biological functions of autophagy genes: a disease perspective. Cell 2019;176:11-42.ArticlePubMedPMC
- 38. Liu M, Huang Y, Xu X, Li X, Alam M, Arunagiri A, et al. Normal and defective pathways in biogenesis and maintenance of the insulin storage pool. J Clin Invest 2021;131:e142240.ArticlePubMedPMC
- 39. Uchizono Y, Alarcon C, Wicksteed BL, Marsh BJ, Rhodes CJ. The balance between proinsulin biosynthesis and insulin secretion: where can imbalance lead? Diabetes Obes Metab 2007;9 Suppl 2:56-66.ArticlePubMed
 PubReader
PubReader ePub Link
ePub Link Cite
Cite