- Current
- Browse
- Collections
-
For contributors
- For Authors
- Instructions to authors
- Article processing charge
- e-submission
- For Reviewers
- Instructions for reviewers
- How to become a reviewer
- Best reviewers
- For Readers
- Readership
- Subscription
- Permission guidelines
- About
- Editorial policy
Articles
- Page Path
- HOME > Diabetes Metab J > Volume 45(1); 2021 > Article
-
Original ArticleGenetics Metagenomic Analysis of the Gut Microbiome Reveals Enrichment of Menaquinones (Vitamin K2) Pathway in Diabetes Mellitus
-
Nihar Ranjan Dash1
 , Mohammad Tahseen Al Bataineh1,2
, Mohammad Tahseen Al Bataineh1,2 -
Diabetes & Metabolism Journal 2021;45(1):77-85.
DOI: https://doi.org/10.4093/dmj.2019.0202
Published online: May 11, 2020
1Department of Clinical Sciences, College of Medicine, University of Sharjah, Sharjah, United Arab Emirates
2Research Institute for Medical & Health Sciences at University of Sharjah, Sharjah, United Arab Emirates
-
Corresponding author: Mohammad Tahseen Al Bataineh Department of Clinical Sciences, College of Medicine, University of Sharjah, 27272 Sharjah, United Arab Emirates. malbataineh@sharjah.ac.ae
Copyright © 2021 Korean Diabetes Association
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
-
Background
- Type 2 diabetes mellitus (T2DM) is a chronic metabolic disease with a high prevalence worldwide, especially among overweight and obese populations. T2DM is multifactorial with several genetic and acquired risk factors that lead to insulin resistance. Mounting evidence indicates that alteration of gut microbiome composition contribute to insulin resistance and inflammation. However, the precise link between T2DM and gut microbiome role and composition remains unknown.
-
Methods
- We evaluated the metabolic capabilities of the gut microbiome of twelve T2DM and six healthy individuals through shotgun metagenomics using MiSeq platform.
-
Results
- We identified no significant differences in the overall taxonomic composition between healthy and T2DM subjects when controlling for differences in diet. However, results showed that T2DM enriched in metabolic pathways involved in menaquinone (vitamin K2) superpathway biosynthesis (PWY-5838) as compared to healthy individuals. Covariance analysis between the bacterial genera and metabolic pathways displaying difference in abundance (analysis of variance P<0.05) in T2DM as compared to healthy subjects revealed that genera belonging Firmicutes, Actinobacteria, and Bacteroidetes phyla contribute significantly to vitamin K2 biosynthesis. Further, the microbiome corresponding to T2DM with high glycosylated hemoglobin (HbA1c) (>6.5%) exhibit high abundance of genes involved in lysine biosynthesis and low abundance of genes involved in creatinine degradation as compared to T2DM with lower HbA1c (<6.5%).
-
Conclusion
- The identified differences in metabolic capabilities provide important information that may eventually lead to the development of novel biomarkers and more effective management strategies to treat T2DM.
- Gut microbiome plays significant role in several cardiometabolic disorders including type 2 diabetes mellitus (T2DM) [1]. Alteration in gut microbiome composition commonly referred as dysbiosis and its consequential functional disturbances attributed to pathogenesis, insulin resistance and treatment response in diabetic individuals [23]. Recent studies have suggested that an altered gut microbiome and the metabolites derived from microbes can alter intestinal barrier, signaling pathways, modulate host physiology and metabolism that directly or indirectly contributes to diabetic states in the host [45].
- In order to understand the role of microbiome on host metabolism and disease, it becomes essential to determine the functional attributes of the microbiome [6]. The metabolic capacity of the microbiome can either be inferred using 16S libraries correlating the phylogenetic trees and clusters of genes shared between taxa [7]. Alternatively, cataloged using shotgun metagenomics libraries providing a direct assessment of the functional attributes of the microbiome depending on sequencing depth [8]. Several studies have reflected on the mechanisms by which microbiome affects the host physiology, assessed from a gene content/functional perspective. For example, it is evident that gut microbiome synthesizes short chain fatty acids, including butyrate, acetate, and propionate. These have been found to be critical for enterocyte homeostasis, and their imbalance has been documented in diseases such like T2DM and inflammatory bowel disease [9]. Similarly, both inflammatory bowel disease and obesity are associated with enrichment of enzymes in the nitrate reductase pathway. Obesity also affects the metabolism of choline and p-cresol, as well as the phosphotransferase system, required for assimilation of dietary carbohydrates [10].
- With the advancements in metagenomics approaches, there is a growing interest in exploring the functionality of the microbiome populations through metabolic pathways and products to better understand the interplay between microbiome and host physiology. While most studies have rallied around short chain fatty acids, enzymes, gaseous products including carbon dioxide, hydrogen, methane, and hydrogen sulfide [11], little is known about vitamins and microbiome micronutrients. Vitamin K2 (also known as menaquinone) synthesized by gut microbiome and found in meat, eggs, curd, cheese, and soybeans is known to play significant role in osteoporosis and cardiovascular diseases [1213]. There are some suggestions that vitamin K2 may be playing some role in improving insulin resistance and reducing T2DM risk through its anti-inflammatory properties, lipid-lowering effects, and the involvement of vitamin K-dependent-protein osteocalcin [1415]. However, the precise link between gut microbiome composition, involved metabolic pathways and vitamin K2 synthesis in T2DM remains unknown. In this study, we explored the metabolic capabilities of the gut microbiome among T2DM and healthy individuals through shotgun metagenomics using MiSeq platform (Illumina, San Diego, CA, USA).
INTRODUCTION
- Ethical statement
- Ethical approval was obtained from University Hospital Sharjah-Hospital Ethics Research Committee (UHS-HERC-021-07022018). All subjects were recruited at University Hospital Sharjah (Sharjah, UAE) and written informed consent was obtained.
- Stool sample collection and preparation
- We collected 18 stool specimens from adult Emirati citizens. Of the 18 subjects, 12 were T2DM and six were healthy subjects. We recruited patient chaperons accompanying T2DM patients during their hospital visits, labeled as healthy control. We determined their health status of controls using a brief medical history, vital signs, and no existing co-morbidities such as T2DM. The basic demographic information such as age, gender, marital status, education, diet, exercise, height, and weight were documented. Glycosylated hemoglobin (HbA1c) measurements were recorded from the patient electronic files. From each subject, 2 to 4 g of freshly passed stool specimen was collected in a sterile container stored immediately into liquid nitrogen and then transferred to −80℃ for further analysis. Liquid (diarrheal) stool and use of antibiotics or probiotics in the last 3 months among the volunteers were the only exclusion criteria used in this study.
- Experimental design
- In order to detect differences in gene potential between healthy and diabetic subjects we sub stratified the samples based on their diets (high and low fiber) calculated using dietary fiber intake short food frequency questionnaire (DFI-FFQ) [16], and HbA1c (marker of average level of blood sugar over the past 3 months) data as shown in Supplementary Table 1.
- DNA extraction and library preparation
- Faecal samples were subjected to DNA extraction using QIAamp PowerFecal DNA kit (Qiagen Ltd., Hilden, Germany) following the manufacturer's instruction. DNA quality was evaluated visually via gel electrophoresis and quantified using a Qubit 3.0 fluorometer (Thermo-Fischer, Waltham, MA, USA). Libraries were prepared using an Illumina Nextera library preparation kit following the standard protocol.
- Sequence technology and processing
- Sequencing was done using an Illumina NextSeq. Around 31.6 Gbases were generated using 2×150 paired-end reads. Each sample yielded a median of 1.8 Gbases. After sequencing, reads were separated according to the barcode used in the library preparation. Initial quality evaluation was done using FastQC v0.11.5. Processing took part in three steps: Paired-ends read joining, removing of contaminants, and trimming. Paired-end reads were joined using FLASH v1.2.11 [17]. Reads were then compared to the Human Genome (hg19, GRCh37 Genome Reference Consortium Human Reference 37) and sequences that mapped to it were removed. Finally sequences were trimmed according to their quality values using Trimmomatic v0.36 [18] using custom parameters (LEADING:5 TRAILING: 5 SLIDINGWINDOW:4:15 MINLEN:70). Joining the paired reads reduced the library size in average by 41.78%. A 0.06% of the stitched reads mapped to the human genome and then removed.
- Statistical analysis
- To evaluate beta diversity across samples, we excluded operational taxonomic units (OTUs) occurring in fewer than 10% of the samples with a count of less than three and calculated Bray-Curtis indices. We tested beta diversity, underscoring differences across samples, using non-metric multidimensional scaling (NMDS) ordination. Dissimilarity in community structure assessed with permutational multivariate analyses of variance (PERMANOVA) with treatment group as the main fixed factor and using 4,999 permutations for significance testing. All analyses conducted in the R environment.
METHODS
- Metagenomic characterization of diabetic and healthy microbiome
- In this study, we conducted a metagenomics characterization of 18 individuals and total bacterial DNA extracted from corresponding fecal samples, then subjected to Illumina shotgun sequencing. Recovered read pools ranged from 14,554,254 to 6,945,034. At the end of quality control, median number of quality-filtered reads per sample was 10,164,901 visualized in Supplementary Fig. 1. These data then used to generate metabolic pathway profiles in the analyzed microbiome utilizing a custom script based on the MetaCyc database [19]. Clinical characteristics of diabetic and healthy individuals were also documented. In order to identify microbial biomarkers mainly attributed to T2DM, we selected individuals with uniform age, gender, ethnicity, diet, and BMI (Supplementary Table 2). Taxonomic composition was determined using Metaphlan2 [20]. The community dominated by bacteria, which accounted in average for 99.051% of the total community followed by archaea, which accounted for 0.863%. On the bacterial side, Firmicutes, Bacteroidetes, and Actinobacteria, which accounted on average for 42.54%, 27.02%, and 20.43% of the bacterial community respectively, dominated the samples. The most common Eukaryotes was Saccharomyces cerevisiae, which accounted for 1e-04% (Supplementary Fig. 2). Firmicutes and Bacteroidetes-dominated microbiome of the adults in this study is consistent with and representative of examined microbiome metabolic profiles in previous reports (Supplementary Fig. 3) [2122].
- Community composition: visualizing similarity among microbiome
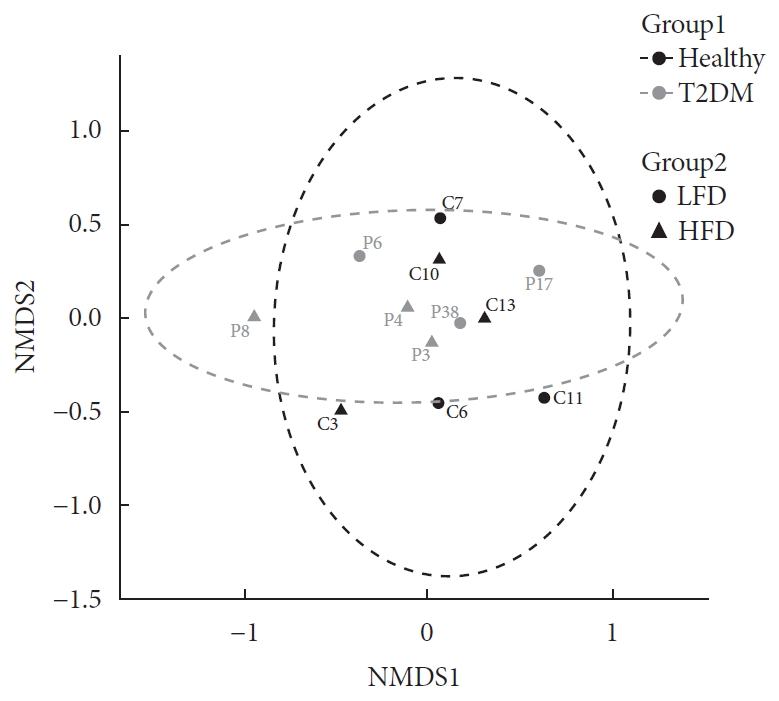
- In order to visualize similarity among microbiome, we examined community compositions. To obtain a graphical representation of similarity among microbial communities, we used an ordination approach. The similarity between any two samples, according to their microbiome composition calculated using Bray-Curtis dissimilarities, a measure that considers both presence/absence and abundance of the species. The distances then evaluated and represented graphically using NMDS ordination (Fig. 1).
- Based on PERMANOVA (using the adonis R function) determined the significance of differences between Group 1 and Group 2, none of the experimental factors (Group 1 and Group 2) or their interaction explained significantly the variation of taxonomic composition profiles. Next, differential abundance testing using DESeq2 (R package) evaluated the distribution of bacterial species between health status when controlling for differences in diet. No bacterial species found to be differentially abundant between the healthy and diabetic patients. Likewise, there was no difference in bacterial species abundance in T2DM with high HbA1c (>6.5%) and low HbA1c (<6.5%), data not shown.
- Functional profiles
- To elucidate functional diversity among groups, we summarized functional profiles into pathways using the Metacyc pathway definition [19]. Around 63.54% reads then mapped into functional genes. Difference in pathway richness (number of unique pathways) then calculated using a generalized linear model assuming and deemed significant. The health status was the only factor that explained significant (P<0.001) differences in richness among patients. This is important to highlight as we normalized for diet as explained before in Supplementary Table 1. Diabetic patients had higher richness of functional genes than healthy subjects did (Fig. 2).
- Furthermore, we analyzed variation of functional groups using PERMANOVA (using the adonis R function) to determine the significance of differences between healthy and T2DM. Interestingly and in contrast to the taxonomic profiles similarity in Fig. 1, one experimental factor (T2DM vs. healthy status) explained significantly part of the variation in functional gene profiles at approximately 21%.
- Differential abundance testing of functional groups
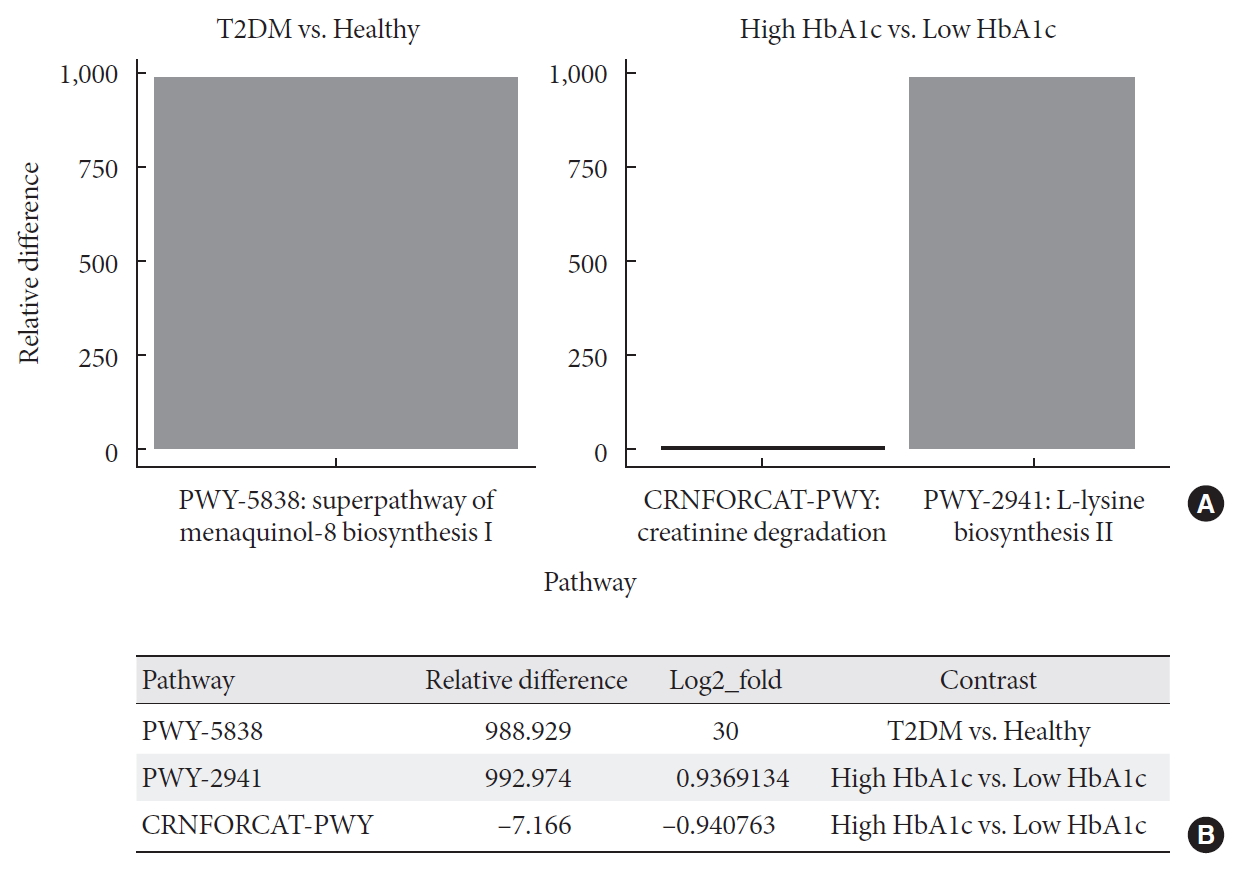
- Metagenome-wide association studies (MGWAS) developed to characterize gut microbiome in human disease, including T2DM and identified biomarkers that can help predict disease occurrence and possibly management outcomes [2324]. Here, we conducted a differential abundance testing of identified functional groups between T2DM, T2DM with high HbA1c, and healthy individuals. Differential abundance testing using DESeq2 (R package) and functional profiles using the Metacyc pathway, summarized into three pathways that significantly differed among healthy, diabetic, and diabetic with high levels of HbA1c subjects (when controlling for differences in diet; P<0.001) (Fig. 3). The table shows log-2-fold differences when compared the differentially abundant pathways between healthy, diabetic, and diabetic with high HbA1c subjects. These pathways are super pathway of menaquinol-8 biosynthesis I (vitamin K2) (PWY-5838) at 30 log-2-fold differences, L-lysine biosynthesis II (lysine acetylation) (PWY-2941) at approximately one log-2-fold differences, and creatinine degradation I at approximately one log-2-fold differences (Fig. 3B).
- Covariance network of bacterial genera and metabolic pathways
- Furthermore, we evaluated covariance between the bacterial genera and metabolic pathways displaying difference in abundance (analysis of variance P<0.05) in T2DM as compared to healthy subjects. Force-driven network representation of these data revealed that genera belonging Firmicutes phylum (10 covariances, P<0.05), Actinobacteria (five covariances, P<0.05), and the Bacteroidetes (five covariances, P<0.05) utilize a key role in modulating vitamin K2 metabolic functionality of the gut microbiome that are altered during T2DM (Fig. 4). Erysipelotrichaceae belongs to Firmicutes phylum representing four out of 10 covariances correlates significantly vitamin K2 pathway (PWY-5838) enrichment in diabetic subjects (Fig. 4). Moreover, Corynebacterium is another gram-positive bacterium that belongs to Actinobacteria phylum represents three out of 10 covariances correlates significantly with vitamin K2 pathway [25] (PWY-5838) enrichment in diabetic subjects (Fig. 4).
RESULTS
- In this study, we report a shift in gut microbiome function among individuals affected by T2DM as compared to healthy controls. Specific profiles of gut microbiome have been linked to T2DM across the globe based on variation in diet, probiotic use, and ethnicity among others [2627]. To overcome this issue, we normalized for fiber diet content, probiotic use, ethnicity and BMI in our study subjects to profile metabolic capabilities of microbiome corresponding to T2DM and healthy individuals. Interestingly, while exploring the community composition among microbiome, we did not detect significant differential abundance between the healthy and diabetic patients. These unexpected results perhaps attributed to small sample size in our study, despite high sequencing counts at an average of 10,164,901 quality-filtered reads per sample. Moreover, one of the potential confounders of our study population was the notable age difference between T2DM and healthy subjects (Supplementary Table 2), that may have an effect over microbiome composition and metabolic profiles that warrants future investigation [28]. Similarly, while we matched gender for our test group (T2DM), we were limited in our gender selection for the control group. We believe gender-specific differences in gut microbiome composition can also be a possible confounder [29]. Additionally, all T2DM individuals in our study population were under Dipeptidyl peptidase-4 inhibitors and metformin treatment that can modulate and affect their microbial diversity [30].
- In contrast to what has previously been reported regarding decrease in the gut microbiome biodiversity with T2DM, we showed higher functional diversity (richness of unique pathways) among T2DM group. Our results suggest an important functional role for the altered microbiome to support their host against metabolic and inflammatory derangement of diabetes (Fig. 2). This gene enrichment in diabetic patients does not necessarily mean a healthy microbiome, but denotes a higher metabolic capability during low-grade inflammation [3132].
- In this study, we characterized for the first time on a possible microbiome micronutrients mechanism to improve host insulin resistance and inflammation. We unveiled enrichment of super pathway of menaquinol-8 biosynthesis I (vitamin K2) (PWY-5838) at 30 log-2-fold differences when compared differential abundance between healthy and diabetic subjects, suggesting an important role of this pathway in insulin resistance and inflammation (Fig. 3).
- Menaquinones (vitamin K2) are considered essential for humans and usually supplemented from nutrient sources and gut bacteria such as Escherichia coli, Bacteroides species, and some gram-positive bacteria [3334]. Previous observational and interventional studies have suggested a key role for vitamin K2 improving insulin resistance among other anti-inflammatory properties such as suppressing nuclear factor κB (NF-κB) signaling pathway [141535363738]. A prospective cohort study following up 38,094 individuals over 10 years concluded that menaquinones intake is associated with reduced risk of T2DM [14]. Others have suggested that vitamin K2 supplementation for few weeks affects β-cell function and/or insulin resistance in healthy subjects [1538]. That said, little is known about the direct link between gut microbiome composition and their contributions to menaquinones production in response to inflammatory conditions such as diabetes.
- Interestingly, identification of highly immunogenic taxa Erysipelotrichaceae and Corynebacterium as a biomarker of T2DM, underscores that changes in relative abundance of certain taxa in the presence of a gut disorder may not reflect a taxa-specific functional role. Erysipelotrichaceae is a gram-positive bacterium that considered highly immunogenic and implicated in inflammatory bowel diseases [394041]. In addition, and consistent with our data shown in Supplementary Table 1, higher levels of Erysipelotrichaceae with obese individuals have been documented elsewhere [42]. Here, we report an important role of Erysipelotrichaceae and Corynebacterium in vitamin K2 production in T2DM subjects. These finding might be useful for new diabetes management strategies to improve insulin resistance.
- In addition, we identified augmentation in L-lysine biosynthesis II (lysine acetylation) (PWY-2941) with high HbA1c T2DM group at approximately one log-2-fold differences when compared differential abundance between low and high HbA1c diabetic subjects, suggesting an important role the gut microbiome in preventing long-term complications of diabetes [43]. In contrast, creatinine degradation I was suppressed at approximately one log-2-fold differences among high HbA1c subjects when compared with low HbA1c diabetics, presumably as a result of altered gut microbiome failing to facilitate creatinine clearance in more complicated diabetic subjects (high HbA1c) [44].
- In conclusion, one can hypothesize that manipulating the intestinal microbiome profiles of especially at-risk individuals might help them to avoid T2DM and other metabolic disorders. We hope that future functional studies will shed more light on microbiome micronutrient contribution to host health and disease conditions.
DISCUSSION
-
CONFLICTS OF INTEREST
This work supported by Research Institute of Medical and Health Sciences at University of Sharjah grant P1701090226 and Boehringer Ingelheim grant 2016-17. The funders stated above had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
-
AUTHOR CONTRIBUTIONS
Conception or design: N.R.D., M.T.A.B.
Acquisition, analysis, or interpretation of data: N.R.D., M.T.A.B.
Drafting the work or revising: N.R.D., M.T.A.B.
Final approval of the manuscript: N.R.D., M.T.A.B.
-
FUNDING
None
NOTES
-
Acknowledgements
- None
ACKNOWLEDGMENTS
SUPPLEMENTARY MATERIALS
Supplementary Table 1
Supplementary Fig. 1
Supplementary Fig. 2
Supplementary Fig. 3
- 1. Tilg H, Moschen AR. Microbiota and diabetes: an evolving relationship. Gut 2014;63:1513-1521.ArticlePubMed
- 2. Larsen N, Vogensen FK, van den Berg FW, Nielsen DS, Andreasen AS, Pedersen BK, et al. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS One 2010;5:e9085.ArticlePubMedPMC
- 3. Zhang X, Shen D, Fang Z, Jie Z, Qiu X, Zhang C, et al. Human gut microbiota changes reveal the progression of glucose intolerance. PLoS One 2013;8:e71108.ArticlePubMedPMC
- 4. Horton F, Wright J, Smith L, Hinton PJ, Robertson MD. Increased intestinal permeability to oral chromium (51 Cr): EDTA in human type 2 diabetes. Diabet Med 2014;31:559-563.ArticlePubMed
- 5. Cani PD, Geurts L, Matamoros S, Plovier H, Duparc T. Glucose metabolism: focus on gut microbiota, the endocannabinoid system and beyond. Diabetes Metab 2014;40:246-257.ArticlePubMed
- 6. Joice R, Yasuda K, Shafquat A, Morgan XC, Huttenhower C. Determining microbial products and identifying molecular targets in the human microbiome. Cell Metab 2014;20:731-741.ArticlePubMedPMC
- 7. Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 2013;31:814-821.ArticlePubMedPMCPDF
- 8. Knight R, Jansson J, Field D, Fierer N, Desai N, Fuhrman JA, et al. Unlocking the potential of metagenomics through replicated experimental design. Nat Biotechnol 2012;30:513-520.ArticlePubMedPMCPDF
- 9. Vital M, Howe AC, Tiedje JM. Revealing the bacterial butyrate synthesis pathways by analyzing (meta)genomic data. mBio 2014;5:e00889.ArticlePubMedPMCPDF
- 10. Levy R, Borenstein E. Metagenomic systems biology and metabolic modeling of the human microbiome: from species composition to community assembly rules. Gut Microbes 2014;5:265-270.ArticlePubMedPMC
- 11. Pimentel M, Mathur R, Chang C. Gas and the microbiome. Curr Gastroenterol Rep 2013;15:356.ArticlePubMedPDF
- 12. Gundberg CM, Markowitz ME, Mizruchi M, Rosen JF. Osteocalcin in human serum: a circadian rhythm. J Clin Endocrinol Metab 1985;60:736-739.ArticlePubMedPDF
- 13. Knapen MH, Braam LA, Drummen NE, Bekers O, Hoeks AP, Vermeer C. Menaquinone-7 supplementation improves arterial stiffness in healthy postmenopausal women: a double-blind randomised clinical trial. Thromb Haemost 2015;113:1135-1144.ArticlePubMedPDF
- 14. Beulens JW, van der A DL, Grobbee DE, Sluijs I, Spijkerman AM, van der Schouw YT. Dietary phylloquinone and menaquinones intakes and risk of type 2 diabetes. Diabetes Care 2010;33:1699-1705.ArticlePubMedPMCPDF
- 15. Choi HJ, Yu J, Choi H, An JH, Kim SW, Park KS, et al. Vitamin K2 supplementation improves insulin sensitivity via osteocalcin metabolism: a placebo-controlled trial. Diabetes Care 2011;34:e147.ArticlePubMedPMCPDF
- 16. Healey G, Brough L, Murphy R, Hedderley D, Butts C, Coad J. Validity and reproducibility of a habitual dietary fibre intake short food frequency questionnaire. Nutrients 2016;8:E558.Article
- 17. Magoc T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011;27:2957-2963.ArticlePubMedPMCPDF
- 18. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014;30:2114-2120.ArticlePubMedPMCPDF
- 19. Caspi R, Billington R, Ferrer L, Foerster H, Fulcher CA, Keseler IM, et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res 2016;44:D471-D480.ArticlePubMedPDF
- 20. Truong DT, Franzosa EA, Tickle TL, Scholz M, Weingart G, Pasolli E, et al. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat Methods 2015;12:902-903.ArticlePubMedPDF
- 21. Claesson MJ, Jeffery IB, Conde S, Power SE, O'Connor EM, Cusack S, et al. Gut microbiota composition correlates with diet and health in the elderly. Nature 2012;488:178-184.ArticlePubMedPDF
- 22. Saraswati S, Sitaraman R. Aging and the human gut microbiota-from correlation to causality. Front Microbiol 2015;5:764.ArticlePubMedPMC
- 23. Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012;490:55-60.ArticlePubMedPDF
- 24. Wang J, Jia H. Metagenome-wide association studies: fine-mining the microbiome. Nat Rev Microbiol 2016;14:508-522.ArticlePubMedPDF
- 25. Abul-Hajj YJ. Isolation of vitamin K2(35) from Nocardia restrictus and Corynebacterium simplex: a natural electron acceptor in microbial steroid ring A dehydrogenations. J Biol Chem 1978;253:2356-2360.ArticlePubMed
- 26. Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, et al. Human gut microbiome viewed across age and geography. Nature 2012;486:222-227.ArticlePubMedPMCPDF
- 27. Tremaroli V, Backhed F. Functional interactions between the gut microbiota and host metabolism. Nature 2012;489:242-249.ArticlePubMedPDF
- 28. An R, Wilms E, Masclee AAM, Smidt H, Zoetendal EG, Jonkers D. Age-dependent changes in GI physiology and microbiota: time to reconsider. Gut 2018;67:2213-2222.ArticlePubMed
- 29. Fransen F, van Beek AA, Borghuis T, Meijer B, Hugenholtz F, van der Gaast-de Jongh C, et al. The impact of gut microbiota on gender-specific differences in immunity. Front Immunol 2017;8:754.ArticlePubMedPMC
- 30. Forslund K, Hildebrand F, Nielsen T, Falony G, Le Chatelier E, Sunagawa S, et al. MetaHIT consortium. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature 2015;528:262-266.ArticlePubMedPMCPDF
- 31. Cani PD, Osto M, Geurts L, Everard A. Involvement of gut microbiota in the development of low-grade inflammation and type 2 diabetes associated with obesity. Gut Microbes 2012;3:279-288.ArticlePubMedPMC
- 32. Chassaing B, Gewirtz AT. Gut microbiota, low-grade inflammation, and metabolic syndrome. Toxicol Pathol 2014;42:49-53.ArticlePubMedPDF
- 33. Conly JM, Stein K. The production of menaquinones (vitamin K2) by intestinal bacteria and their role in maintaining coagulation homeostasis. Prog Food Nutr Sci 1992;16:307-343.PubMed
- 34. Suttie JW. The importance of menaquinones in human nutrition. Annu Rev Nutr 1995;15:399-417.ArticlePubMed
- 35. Yoshida M, Jacques PF, Meigs JB, Saltzman E, Shea MK, Gundberg C, et al. Effect of vitamin K supplementation on insulin resistance in older men and women. Diabetes Care 2008;31:2092-2096.ArticlePubMedPMCPDF
- 36. Yamaguchi M, Weitzmann MN. Vitamin K2 stimulates osteoblastogenesis and suppresses osteoclastogenesis by suppressing NF-κB activation. Int J Mol Med 2011;27:3-14.ArticlePubMed
- 37. Thakore AH, Guo CY, Larson MG, Corey D, Wang TJ, Vasan RS, et al. Association of multiple inflammatory markers with carotid intimal medial thickness and stenosis (from the Framingham Heart Study). Am J Cardiol 2007;99:1598-1602.ArticlePubMed
- 38. Sakamoto N, Nishiike T, Iguchi H, Sakamoto K. Possible effects of one week vitamin K (menaquinone-4) tablets intake on glucose tolerance in healthy young male volunteers with different descarboxy prothrombin levels. Clin Nutr 2000;19:259-263.ArticlePubMed
- 39. Palm NW, de Zoete MR, Cullen TW, Barry NA, Stefanowski J, Hao L, et al. Immunoglobulin A coating identifies colitogenic bacteria in inflammatory bowel disease. Cell 2014;158:1000-1010.ArticlePubMedPMC
- 40. Dinh DM, Volpe GE, Duffalo C, Bhalchandra S, Tai AK, Kane AV, et al. Intestinal microbiota, microbial translocation, and systemic inflammation in chronic HIV infection. J Infect Dis 2015;211:19-27.ArticlePubMedPDF
- 41. Schaubeck M, Clavel T, Calasan J, Lagkouvardos I, Haange SB, Jehmlich N, et al. Dysbiotic gut microbiota causes transmissible Crohn's disease-like ileitis independent of failure in antimicrobial defence. Gut 2016;65:225-237.ArticlePubMed
- 42. Zhang H, DiBaise JK, Zuccolo A, Kudrna D, Braidotti M, Yu Y, et al. Human gut microbiota in obesity and after gastric bypass. Proc Natl Acad Sci U S A 2009;106:2365-2370.ArticlePubMedPMC
- 43. Kosanam H, Thai K, Zhang Y, Advani A, Connelly KA, Diamandis EP, et al. Diabetes induces lysine acetylation of intermediary metabolism enzymes in the kidney. Diabetes 2014;63:2432-2439.ArticlePubMedPDF
- 44. Jones JD, Burnett PC. Creatinine metabolism in humans with decreased renal function: creatinine deficit. Clin Chem 1974;20:1204-1212.ArticlePubMedPDF
REFERENCES
Evaluation of community composition among samples. Panel shows non-metric multidimensional scaling (NMDS) ordination. Ordination plots were categorized by color according to the patient health status (Group 1) and by shape according to their diet (Group 2). T2DM, type 2 diabetes mellitus; LFD, low fiber diet; HFD, high fiber diet.
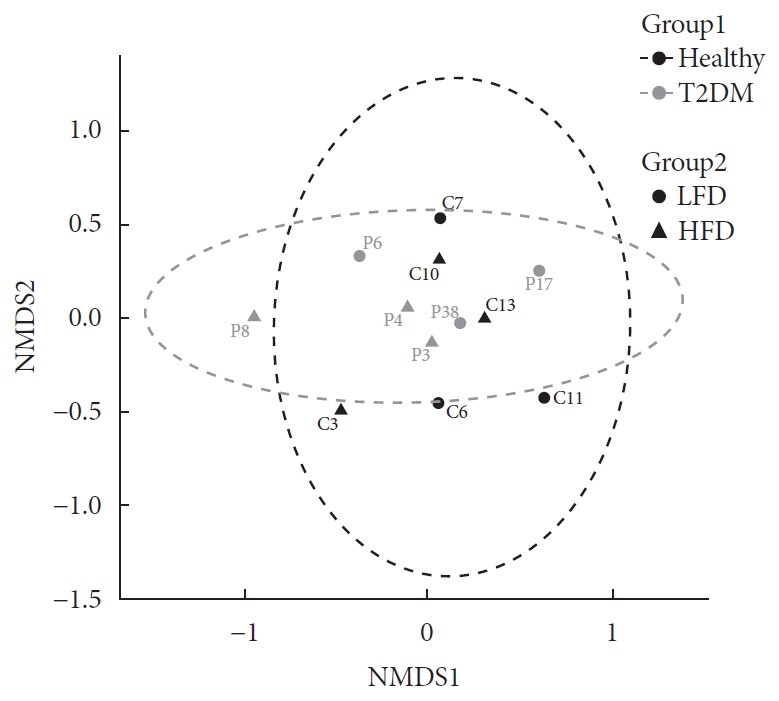
Difference of functional profiles among samples. Panel shows difference in pathway richness (number of unique pathways) for (A) healthy and (B) type 2 diabetes mellitus (T2DM). Diabetic patients had higher richness of functional genes than that of healthy subjects (P<0.001) irrespective to differences in diet. LFD, low fiber diet; HFD, high fiber diet.
Differential abundance testing of functional groups. (A) Panel shows pathways with significant relative abundance (P<0.001). (B) Log fold difference for significantly enriched pathways for type 2 diabetes mellitus (T2DM) and T2DM with high glycosylated hemoglobin (HbA1c) are PWY-5838 30 fold, PWY-2941 0.9 fold, and CRNFORCAT-PWY −0.9 fold in contrast to healthy and T2DM with low HbA1c.
Covariance network of bacterial genera and metabolic pathways. Panel a shows a force-driven network based on the predicted covariances (analysis of variance with P<0.05) between the genera and metabolic pathways identified as statistically altered in type 2 diabetes mellitus as compared to healthy.
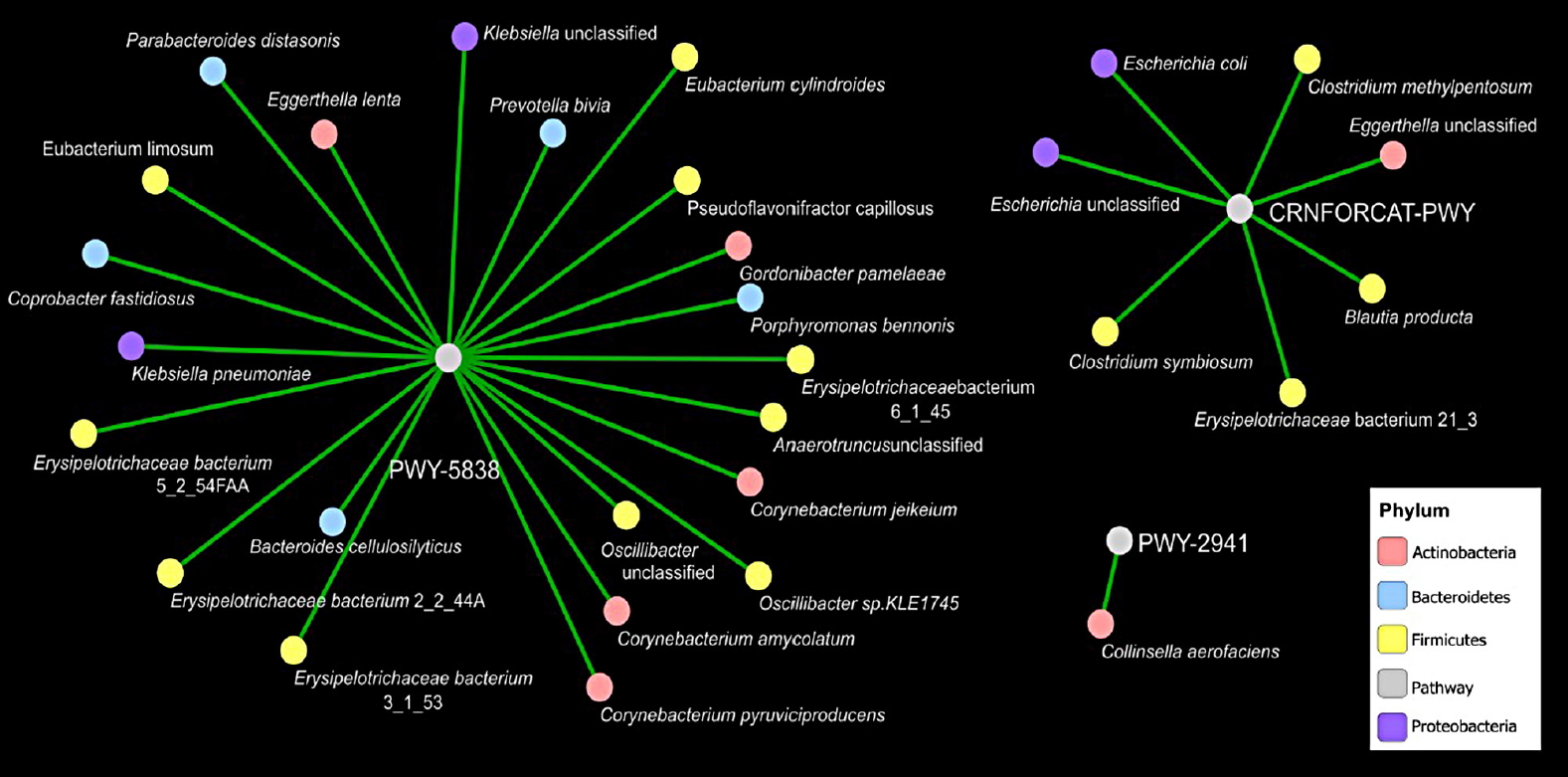
Figure & Data
References
Citations
- Unraveling metagenomics through long-read sequencing: a comprehensive review
Chankyung Kim, Monnat Pongpanich, Thantrira Porntaveetus
Journal of Translational Medicine.2024;[Epub] CrossRef - Longitudinal gut microbiome changes in immune checkpoint blockade-treated advanced melanoma
Johannes R. Björk, Laura A. Bolte, Andrew Maltez Thomas, Karla A. Lee, Niccolo Rossi, Thijs T. Wind, Lotte M. Smit, Federica Armanini, Francesco Asnicar, Aitor Blanco-Miguez, Ruth Board, Neus Calbet-Llopart, Lisa Derosa, Nathalie Dhomen, Kelly Brooks, Mar
Nature Medicine.2024; 30(3): 785. CrossRef - Chimonanthus nitens Oliv leaves essential oil alleviates colitis induced by sodium dextran sulfate in BALB/C mice via MAPK/NF-κB/Nrf2 signaling pathway
Yang Zhang, Jing He, Lingli Chen, Wenjun Wang
Journal of Functional Foods.2024; 115: 106095. CrossRef - The gut microbiota pathway mechanisms of diabetes
Ousman Bajinka, Yurong Tan, Alansana Darboe, Isabella Gloria Ighaede-Edwards, Khalid A. Abdelhalim
AMB Express.2023;[Epub] CrossRef - Adiposity is associated with expansion of the genus Dialister in rheumatoid arthritis patients
Natalia Mena-Vázquez, Patricia Ruiz-Limón, Isabel Moreno-Indias, Sara Manrique-Arija, Jose Manuel Lisbona-Montañez, José Rioja, Arkaitz Mucientes, Gracia María Martin-Núñez, Laura Cano-García, Francisco J. Tinahones, Antonio Fernández-Nebro
Biomedicine & Pharmacotherapy.2023; 160: 114388. CrossRef - Virulence Factors of the Gut Microbiome Are Associated with BMI and Metabolic Blood Parameters in Children with Obesity
S. M. Murga-Garrido, E. J. Ulloa-Pérez, C. E. Díaz-Benítez, Y. C. Orbe-Orihuela, F. Cornejo-Granados, A. Ochoa-Leyva, A. Sanchez-Flores, M. Cruz, A. C. Castañeda-Márquez, T. Plett-Torres, A. I. Burguete García, A. Lagunas-Martínez, Jennifer M. Auchtung, J
Microbiology Spectrum.2023;[Epub] CrossRef - The microbiome in adolescents with irritable bowel syndrome and changes with percutaneous electrical nerve field stimulation
Daniel F. Castillo, Lee A. Denson, David B. Haslam, Kevin A. Hommel, Nicholas J. Ollberding, Rashmi Sahay, Neha R. Santucci
Neurogastroenterology & Motility.2023;[Epub] CrossRef - Uncovering the relationship between gut microbial dysbiosis, metabolomics, and dietary intake in type 2 diabetes mellitus and in healthy volunteers: a multi-omics analysis
Mohammad Tahseen Al Bataineh, Axel Künstner, Nihar Ranjan Dash, Habiba S. Alsafar, Mohab Ragab, Franziska Schmelter, Christian Sina, Hauke Busch, Saleh Mohamed Ibrahim
Scientific Reports.2023;[Epub] CrossRef - The Synergism of Human Lactobacillaceae and Inulin Decrease Hyperglycemia via Regulating the Composition of Gut Microbiota and Metabolic Profiles in db/db Mice
Peifan Li, Tong Tong, Yusong Wu, Xin Zhou, Michael Zhang, Jia Liu, Yongxin She, Zuming Li, Yongli Li
Journal of Microbiology and Biotechnology.2023; 33(12): 1657. CrossRef - Functional alterations and predictive capacity of gut microbiome in type 2 diabetes
Nihar Ranjan Dash, Mohammad T. Al Bataineh, Rohia Alili, Habiba Al Safar, Noura Alkhayyal, Edi Prifti, Jean-Daniel Zucker, Eugeni Belda, Karine Clément
Scientific Reports.2023;[Epub] CrossRef - Schizophyllum commune-derived β-glucan improves intestinal health demonstrating protective effects against constipation and common metabolic disorders
Vuong Vu, Karthika Muthuramalingam, Vineet Singh, Changmin Choi, Young Mee Kim, Tatsuya Unno, Moonjae Cho
Applied Biological Chemistry.2022;[Epub] CrossRef - Effects of Non-Polar Dietary and Endogenous Lipids on Gut Microbiota Alterations: The Role of Lipidomics
Konstantinos Tsiantas, Spyridon J. Konteles, Eftichia Kritsi, Vassilia J. Sinanoglou, Thalia Tsiaka, Panagiotis Zoumpoulakis
International Journal of Molecular Sciences.2022; 23(8): 4070. CrossRef - Vitamin K2 Enhances Fat Degradation to Improve the Survival of C. elegans
Zhi Qu, Lu Zhang, Wei Huang, Shanqing Zheng
Frontiers in Nutrition.2022;[Epub] CrossRef - The Effect of Polyphenol Extract from Rosa Roxburghii Fruit on Plasma Metabolome and Gut Microbiota in Type 2 Diabetic Mice
Hui Wang, Zhaojun Chen, Mei Wang, Mingxiu Long, Tingyuan Ren, Chao Chen, Xiaotong Dai, Sheng Yang, Shuming Tan
Foods.2022; 11(12): 1747. CrossRef - Untargeted Metabolomic Plasma Profiling of Emirati Dialysis Patients with Diabetes versus Non-Diabetic: A Pilot Study
Bayan Hassan Banimfreg, Hussam Alshraideh, Abdulrahim Shamayleh, Adnane Guella, Mohammad Harb Semreen, Mohammad Tahseen Al Bataineh, Nelson C. Soares
Biomolecules.2022; 12(7): 962. CrossRef - Recent Progress in the Diagnosis and Management of Type 2 Diabetes Mellitus in the Era of COVID-19 and Single Cell Multi-Omics Technologies
Krisztina Kupai, Tamás Várkonyi, Szilvia Török, Viktória Gáti, Zsolt Czimmerer, László G. Puskás, Gábor J. Szebeni
Life.2022; 12(8): 1205. CrossRef - Personalized microbiome-driven effects of non-nutritive sweeteners on human glucose tolerance
Jotham Suez, Yotam Cohen, Rafael Valdés-Mas, Uria Mor, Mally Dori-Bachash, Sara Federici, Niv Zmora, Avner Leshem, Melina Heinemann, Raquel Linevsky, Maya Zur, Rotem Ben-Zeev Brik, Aurelie Bukimer, Shimrit Eliyahu-Miller, Alona Metz, Ruthy Fischbein, Olga
Cell.2022; 185(18): 3307. CrossRef - Microbial Communities in Home-Made and Commercial Kefir and Their Hypoglycemic Properties
Birsen Yilmaz, Emine Elibol, H. Nakibapher Jones Shangpliang, Fatih Ozogul, Jyoti Prakash Tamang
Fermentation.2022; 8(11): 590. CrossRef - Microbial vitamin production mediates dietary effects on diabetic risk
Daoming Wang, Van T. Pham, Robert E. Steinert, Alexandra Zhernakova, Jingyuan Fu
Gut Microbes.2022;[Epub] CrossRef - Clinical benefits of β-glucan supplementation in children: a review
Rachana Bhoite, Vinita Satyavrat, Manasa Premasudha Sadananda
Discover Food.2022;[Epub] CrossRef - Vitamin K2 Holds Promise for Alzheimer’s Prevention and Treatment
Alexander Popescu, Monica German
Nutrients.2021; 13(7): 2206. CrossRef - Type 1 diabetes in pregnancy is associated with distinct changes in the composition and function of the gut microbiome
Alexandra J. Roth-Schulze, Megan A. S. Penno, Katrina M. Ngui, Helena Oakey, Esther Bandala-Sanchez, Alannah D. Smith, Theo R. Allnutt, Rebecca L. Thomson, Peter J. Vuillermin, Maria E. Craig, William D. Rawlinson, Elizabeth A. Davis, Mark Harris, Georgia
Microbiome.2021;[Epub] CrossRef
 PubReader
PubReader ePub Link
ePub Link Cite
Cite