- Current
- Browse
- Collections
-
For contributors
- For Authors
- Instructions to authors
- Article processing charge
- e-submission
- For Reviewers
- Instructions for reviewers
- How to become a reviewer
- Best reviewers
- For Readers
- Readership
- Subscription
- Permission guidelines
- About
- Editorial policy
Articles
- Page Path
- HOME > Diabetes Metab J > Volume 44(1); 2020 > Article
-
ReviewBasic Research Mitochondrial Mechanisms in Diabetic Cardiomyopathy
-
Johannes Gollmer
 , Andreas Zirlik, Heiko Bugger
, Andreas Zirlik, Heiko Bugger -
Diabetes & Metabolism Journal 2020;44(1):33-53.
DOI: https://doi.org/10.4093/dmj.2019.0185
Published online: February 21, 2020
Division of Cardiology, Medical University of Graz, Graz, Austria.
- Corresponding author: Heiko Bugger. Division of Cardiology, Medical University of Graz, Auenbruggerplatz 15, 8036 Graz, Austria. heiko.bugger@medunigraz.at
• Received: October 15, 2019 • Accepted: December 20, 2019
Copyright © 2020 Korean Diabetes Association
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- Mitochondrial medicine is increasingly discussed as a promising therapeutic approach, given that mitochondrial defects are thought to contribute to many prevalent diseases and their complications. In individuals with diabetes mellitus (DM), defects in mitochondrial structure and function occur in many organs throughout the body, contributing both to the pathogenesis of DM and complications of DM. Diabetic cardiomyopathy (DbCM) is increasingly recognized as an underlying cause of increased heart failure in DM, and several mitochondrial mechanisms have been proposed to contribute to the development of DbCM. Well established mechanisms include myocardial energy depletion due to impaired adenosine triphosphate (ATP) synthesis and mitochondrial uncoupling, and increased mitochondrial oxidative stress. A variety of upstream mechanisms of impaired ATP regeneration and increased mitochondrial reactive oxygen species have been proposed, and recent studies now also suggest alterations in mitochondrial dynamics and autophagy, impaired mitochondrial Ca2+ uptake, decreased cardiac adiponectin action, increased O-GlcNAcylation, and impaired activity of sirtuins to contribute to mitochondrial defects in DbCM, among others. In the current review, we present and discuss the evidence that underlies both established and recently proposed mechanisms that are thought to contribute to mitochondrial dysfunction in DbCM.
- Despite a risen awareness among the clinical society, diabetes mellitus (DM) remains a growing epidemic which affected 425 million adults in 2017, and this number is expected to increase to 629 million individuals affected by DM until 2045 (www.idf.org). DM has a huge impact on the prognosis of these patients through the traditional accompanying macro- and microvascular complications which are leading to a markedly increased morbidity and mortality. Since the Framingham Heart Study, it is known that DM also increases the incidence of heart failure (HF), which is mainly related to accelerated atherosclerosis and an increased incidence and severity of myocardial infarction, as well as the frequent coexistence of arterial hypertension in diabetic individuals. However, the risk for HF is also increased in an underestimated amount of diabetic patients in the absence of these risk factors, an entity termed diabetic cardiomyopathy (DbCM) [1]. Given that many molecular alterations and mechanisms identified in failing hearts are also similarly altered in DbCM, DbCM can be considered a predisposition to develop cardiac dysfunction, in particular in the presence of other stress factors such as ischemic heart disease or hypertension. Mitochondria are the powerhouse of the cell, continuously providing large amounts of adenosine triphosphate (ATP) to cardiomyocytes which require an extraordinary ATP turnover to maintain contractile function. Of note, not only in HF but also in DbCM, a number of defects in mitochondrial biology have been consistently reported, and a variety of distinct mechanisms have been proposed to lead to these mitochondrial defects [2]. Nevertheless, the underlying mechanisms of mitochondrial defects remain incompletely understood, and with increasing understanding of mitochondrial biology in recent years, a number of novel pathways and mechanisms have been elucidated that may contribute to mitochondrial dysfunction in the diabetic heart. In the current review, we will discuss established and novel mechanisms of myocardial mitochondrial dysfunction in DM.
INTRODUCTION
- The first evidence supporting the existence of a DbCM has been published by Rubler et al. [1] in 1972 who identified four patients in a post-mortem analysis that died from HF, had suffered from DM, but had no other known etiology of HF. Subsequently, the Framingham Heart Study reported a significantly increased incidence of HF in patients with DM, with a 2-fold increase in men and a 5-fold increase in women [3]. Besides many more studies confirming the increased risk for HF in diabetics, DM has also been shown to be over-represented in patients hospitalized for HF, and the prognosis of HF in type 2 diabetes mellitus (T2DM) patients is worse than in patients with HF alone [4567]. These observations have led to the hypothesis that diabetic patients may develop molecular alterations affecting the heart independently of the macrovascular complications like coronary artery disease (CAD) or hypertension which are also driven by DM [8]. Today, the definition of DbCM describes a cardiac dysfunction in diabetic individuals in the absence of CAD, hypertension, valvular heart disease, congenital heart disease, or any other known etiology of cardiomyopathy [9].
- The clinical phenotype of DbCM has been a subject of debate. The typical traits of DbCM are thought to include cardiac hypertrophy and diastolic dysfunction, which may become clinically manifest as heart failure with preserved ejection fraction (HFpEF) [910]. In addition, a number of studies reported subclinical systolic dysfunction in patients with DbCM detected by echocardiographic strain analysis. Some authors think of these two phenotypes as one continuum where DbCM progresses from the HFpEF phenotype with cardiac hypertrophy and concomitant diastolic dysfunction to a later stage with further structural damage, leading to a rather dilated phenotype with systolic impairment [11]. Others suppose that the two phenotypes may be different entities with distinct mechanisms contributing to either one [1012]. Longitudinal data observing the evolution of DbCM in a well-defined patient cohort with exclusion of confounding pathologies would be needed to shed more light on this issue.
DEFINITION AND CLINICAL PHENOTYPE OF DbCM
- Numerous mechanisms have been identified and proposed to contribute to the pathogenesis of DbCM, including oxidative stress, fibrosis, inflammation, various forms of cell death, alterations in myocardial energetics, among others [2]. Of particular importance may be the development of mitochondrial dysfunction, which may not only contribute to the development of diabetic complications in different organs, but may also contribute to the development of T2DM by inducing insulin resistance in skeletal muscle, adipose tissue and pancreatic β-cells [13]. In rodents models of DM, which are per se resistant to the development of CAD and hypertension, defects in mitochondrial biology have been observed already several decades ago. As early as 1985, an impairment in state 3 respiration of isolated mitochondria has been reported in hearts of obese or diabetic mice [14]. Ever since, mitochondrial dysfunction has been observed in numerous rodent models of DM, including models of type 1 diabetes mellitus (T1DM) such as streptozotocin (STZ)-diabetic rodents, OVE26 mice, non-obese diabetic (NOD) mice, or in the Akita mouse model, as well as models of T2DM such as ob/ob mice, db/db mice, Zucker (diabetic) fatty rats, Goto Kakizaki rats, and in models of diet-induced obesity [15]. In humans, mitochondrial dysfunction was observed in atrial tissue of DM patients by Anderson et al. [16] who demonstrated impaired respiration rates of isolated mitochondria using fatty acids (FAs) or glutamate as a substrate, and increased generation of hydrogen peroxide (H2O2). Furthermore, studies using atrial tissue or tissue of atrial appendage also reported impaired respiration rates and electron transport chain (ETC) complex activities in diabetic individuals [1718]. Taken together, there is compelling evidence that alterations in mitochondrial function exist in rodent and human DbCM. Underlying mechanisms of impaired mitochondrial biology in DbCM will be discussed in the following sections (Fig. 1).
MITOCHONDRIAL DYSFUNCTION IN DbCM
- Altered mitochondrial substrate utilization
- To maintain continuous pump function, the heart requires large amounts of high energy phosphates and accounts for approximately 8% of the total ATP consumption of the body. The vast majority of this ATP is regenerated in the mitochondria via oxidative phosphorylation (OXPHOS), which explains the high mitochondrial volume density of 30% to 40% in the heart, dependent on the species [19]. In the absence of DM or other cardiac pathologies, the majority of ATP is derived from the oxidation of FAs (60% to 70%), whereas a minor part is derived from the oxidation of glucose, lactate, ketone bodies, and amino acids (20% to 30%), depending on their availability in the blood [20212223]. The resulting reducing equivalents (NADH, FADH2) deliver electrons into the ETC, where electrons are transported through the distinct complexes of the ETC and finally transferred onto molecular oxygen by the activity of complex IV, thereby reducing O2 to H2O. This electron transport is used by the ETC complexes to build up an electrochemical gradient by pumping protons into the intermembranous space. The energy released by back flow of protons into the mitochondrial matrix via the FO subunit of the FOF1-ATPase is used by the FOF1-ATPase to regenerate ATP from adenosine diphosphate (ADP); thus, ATP regeneration is coupled to oxygen consumption.
- In DM, the typically observed increase in serum FAs and triglycerides promotes an increase in FA uptake and oxidation. Evaluation of myocardial substrate oxidation in isolated working hearts demonstrated increased rates of fatty acid oxidation (FAO) and decreased oxidation of glucose in various animal models of T2DM, including db/db mice, ob/ob mice, or Zucker diabetic fatty rats [2425]. Similar observations have been made in humans, where rates of FA uptake and oxidation were increased and insulin-stimulated glucose uptake and glucose utilization were decreased in insulin-resistant and/or diabetic individuals [26272829]. Increased FAO rates are driven, at least in part, by increased activity of peroxisome proliferator-activated receptors (PPARs), in particular PPARα. Both activation of PPARα by FAs and peroxisome proliferator-activated receptor gamma coactivator 1α (PGC-1α) as well as increased PPARα expression increase the binding of PPARα to PPAR response elements on promoters of genes encoding for proteins involved in FA uptake and oxidation, including carnitine palmitoyltransferase 1 (CPT1) and long chain acyl-CoA dehydrogenase (LCAD) as key enzymes of FAO [30]. Indeed, cardiomyocyte-specific overexpression of PPARα resulted in increased expression of FA utilization genes and increased FAO [31]. In turn, expression of genes encoding for enzymes involved in glucose oxidation was reduced, accompanied by decreased glucose uptake and oxidation. Of note though, PPARα signaling was not increased as early as 4 weeks of age in these mice, whereas FAO rates were increased. Thus, PPARα-independent mechanisms may increase FAO rates in the early disease state, whereas PPARα signaling may sustain elevated rates of FAO in the heart as DM persists.
- Increased rates of FAO affect the mitochondrial efficiency of ATP regeneration. Mjos [32] demonstrated more than four decades ago that increasing cardiac FA uptake by lipid infusion leads to enhanced oxygen extraction in healthy dogs. Since cardiac contractility remained unaltered, the ratio of cardiac work to O2 consumption (i.e., cardiac efficiency) was impaired. Similarly, FAO and myocardial O2 consumption are increased and cardiac efficiency is decreased in hearts of ob/ob and db/db mice, as well as in humans with obesity and insulin resistance [242733]. The mechanism of increased O2 consumption may be caused by mitochondrial uncoupling. Boudina et al. [3334] demonstrated that the presence of long chain FA in the perfusion medium during Langendorff perfusion of db/db hearts increased O2 consumption and impaired the ATP/O ratio, indicating FA-induced mitochondrial uncoupling. The proton leak could be inhibited by guanosine triphosphate (GTP), which blocks uncoupling protein (UCP) activity. Given an increase in mitochondrial ROS, it has been proposed that increased mitochondrial ROS would directly activate mitochondrial UCPs, thereby increasing oxygen consumption and impairing cardiac contractility due to decreased ATP regeneration, resulting in impaired cardiac efficiency. A small component of the proton leak in db/db heart mitochondria was also sensitive to inhibition with atractyloside, suggesting some uncoupling to be mediated also by activity of the adenine nucleotide translocase. Of note, mice lacking insulin receptors in cardiomyocytes (CIRKO mice) also showed impaired ATP/O ratios when respiring palmitoyl-carnitine, which was normalized by scavenging mitochondrial ROS using manganese (III) tetrakis (4-benzoic acid) porphyrin (MnTBAP), thereby not only confirming that ROS may induce uncoupling but also suggesting a critical role of cardiac insulin resistance in the pathogenesis of ROS-induced mitochondrial uncoupling [35]. Furthermore, increasing FA delivery to CIRKO hearts by induction of STZ-dependent DM was a prerequisite to develop increased O2 consumption and impaired cardiac efficiency [36]. Given that CIRKO mice displayed marked proteomic remodeling with downregulation of FAO proteins, it was proposed that overwhelming the FAO capacity of CIRKO mice by additional hyperlipidemia may have contributed to induction of uncoupling and cardiac inefficiency. Importantly, hearts of ob/ob and db/db mice display cardiomyocyte insulin resistance when evaluated by insulin-stimulated glucose uptake and glycolysis [37]. Thus, it can be speculated that insulin-resistant cardiomyocytes in T2DM hearts are susceptible to FA-induced ROS-mediated activation of UCPs, thereby impairing cardiac efficiency.
- If the delivery of FA to mitochondria exceeds the mitochondrial capacity for FAO, acyl-CoAs accumulate and are directed into other pathways of lipid metabolism such as synthesis of triacylglycerols (TAG), ceramides, diacylglycerols, and acyl-carnitines [38]. Not only in many animal models of diabetes, but also in human diabetic subjects, diabetes drives accumulation of TAG in the heart [3940]. Recent lipidomics profiling also demonstrated a pronounced remodeling of the myocardial phospholipid pool in rat DbCM [41]. While storage of FA as TAG may serve a protective function to prevent excessive generation of reactive lipid intermediates, ceramide and diacylglycerol accumulation might contribute to the development of lipotoxic cardiac dysfunction, e.g., by activation of protein kinase C signaling, apoptosis, endoplasmic reticulum (ER) stress, and increased ROS generation [124243]. Levels of ceramides can be markedly increased in obese and diabetic rats, and inhibition of the rate-limiting enzyme of ceramide biosynthesis, serine palmitoyltransferase, resulted in improved systolic function in isolated hearts of mice with lipotoxic cardiomyopathy [44]. Accumulation of acyl-CoAs may also be detrimental to the diabetic heart, given that increased cardiomyocyte levels of acyl-CoA in mice overexpressing long-chain acyl-CoA synthetase 1 (ACSL1) show increased apoptosis and ceramide levels, marked fragmentation of mitochondria, increased mitochondrial ROS generation, and defects in cardiac contractility [4546]. While the role of phospholipid remodeling in DbCM remains unresolved, it is tempting to speculate that mitochondrial membrane remodeling could potentially alter OXPHOS function, ROS generation and the process of mitochondrial fusion and fission.
- Another effect of FA on the myocardium in diabetes relates to the cardiac adaptation to hypoxic conditions [47]. Work from the Heather lab showed that diabetic hearts fail to accumulate hypoxia-inducible factor (HIF)-1α during ischemia. Using a model of insulin resistance in HL-1 cardiomyocytes, they showed that long-chain FA may prevent HIF-1α accumulation and subsequent protective HIF-1α downstream signaling due to prevention of succinate accumulation, suggesting that increased FA levels in diabetes may impair cardiac HIF-1α accumulation and subsequent cardioprotection [47].
- A cytokine-related mechanism that may cause mitochondrial dysfunction and uncoupling in diabetic hearts is impaired cardiac adiponectin action. Adiponectin is an adipose-derived cytokine, whose levels are decreased in obese and diabetic individuals, and hypoadiponectinemia has been established as an independent risk factor for cardiovascular disease [48]. Adiponectin deficiency has been shown to impair mitochondrial function in various tissues, and normalizing serum adiponectin levels in ob/ob mice attenuated defects in ETC complex activities [49505152]. These effects of adiponectin are likely mediated via adiponectin receptor 1 (AdipoR1), the main receptor for adiponectin expressed in cardiomyocytes. Similar to adiponectin deficiency, deletion of AdipoR1 in skeletal muscle compromised mitochondrial biogenesis and ETC complex activities by suppressing the signaling axis of AMP-activated protein kinase (AMPK)-sirtuin 1 (SIRT1)-PGC-1α [5354]. In the heart, we demonstrated that deletion of AdipoR1 but not AdipoR2 results in impaired ETC complex activities, accompanied by decreased expression of ETC subunits and impaired AMPK-SIRT1-PGC-1α signaling [55]. We also observed decreased ATP/O ratios and an impairment in cardiac efficiency, and both were normalized by mitochondria-targeted ROS scavenging. In addition, inhibition of UCPs with guanosine diphosphate also normalized mitochondrial uncoupling, indicative of ROS-induced uncoupling. Of note though, FAO rates were not increased, suggesting that ROS-induced UCP-mediated uncoupling and impairment in cardiac efficiency may not depend on FAs in this model. Nevertheless, since both serum adiponectin levels and cardiac AdipoR1 expression are reduced in models of diet-induced obesity and DM, impaired AdipoR1 signaling may also contribute to impaired mitochondrial coupling and cardiac efficiency in DbCM (unpublished results) [51].
- It remains to be mentioned though that not all animal models of DM (e.g., Zucker diabetic fatty [ZDF] rats, T1DM Akita mice) develop increased O2 consumption, mitochondrial uncoupling or impaired cardiac efficiency [56]. In addition, other mechanisms may contribute to impaired cardiac efficiency, such as FAs being a less efficient fuel for ATP regeneration. Theoretical calculations predict that shifting substrate oxidation from 100% palmitate to 100% glucose would increase ATP yield by 12% to 14%, thus suggesting a higher oxygen cost to produce ATP and thereby also explaining increased O2 consumption and impaired cardiac efficiency. However, since the relative substrate shift in diabetic hearts from glucose to FAs is much less pronounced than in this theoretical calculation, such a mechanisms may only become relevant in a setting of increased energy demand, such as marked hypertension or ischemic insults.
- Oxidative stress
- One of the established mechanisms contributing to DbCM is oxidative stress through imbalanced generation and scavenging of ROS. Superoxide (O2·−) can be generated by the reduction of molecular oxygen, is highly reactive and a precursor of other species of ROS. H2O2, being a non-radical form of ROS, can result from dismutation of O2·−, either spontaneously or by the activity of superoxide dismutases (SODs). O2·− and H2O2 can generate the hydroxyl radical (·OH) which may have the greatest oxidative potential of all forms of ROS. In the heart, mitochondria may represent the major source of ROS production. Within mitochondria, the main source of ROS is the ETC, both resulting from unspecific leakage of electrons and also driven by the activity of ETC complexes, mainly complex I and III. At complex I, generation of O2·− occurs primarily at the flavin mononucleotide prosthetic group, whereas ROS generation at complex III seems to occur at the ubisemiquinone which is bound at the Qo-site [57]. Minor amounts of ROS are generated by the other complexes or by reverse electron transport (RET). ETC-independent sources of mitochondrial ROS include monoamine oxidases (MAO) that generate H2O2 during deamination of different neurotransmitters, and NADPH oxidase 4 (NOX4) which produces O2·−. Production of ROS is counterbalanced by ROS detoxification to regulate ROS homeostasis, including a complex antioxidant system within mitochondria. Manganese superoxide dismutase (MnSOD) converts O2·− to H2O2, which can then be reduced to H2O by catalase and/or an antioxidative system comprised of gluthione, gluthathione peroxidases (Gpx), peroxiredoxins (Prx), and thioredoxins (Trx), dependent on the redox status and availability of reducing equivalents within mitochondria [5859]. Non-enzymatic antioxidant mechanisms include cytochrome c and coenzyme Q. Next to a physiologic function in modulating intracellular signaling, increased mitochondrial ROS induce oxidative damage to DNA, proteins and lipids, and may trigger a variety of pathological pathways involved in mitochondrial and cellular damage. Because of their short half-live, it is assumed that ROS predominantly cause harm close to their origin.
- Good evidence exists for increased mitochondrial ROS in rodent and human DbCM [16336061]. Attenuation of mitochondrial ROS by overexpression of catalase, MnSOD or Prx3, or by treatment with mitochondria-targeted antioxidant agents such as Mito-TEMPOL, did attenuate mitochondrial oxidative stress, mitochondrial structural and functional defects, cardiac hypertrophy and contractile dysfunction, thereby establishing the causal link between mitochondrial ROS and development of DbCM [62636465]. Regarding sources and consequences of mitochondrial ROS, incubation of cardiomyocytes from diabetic OVE26 mice in high-glucose medium lead to increased ROS production, which was prevented by inhibition of complex I or II, along with improvement of cardiomyocyte contractility, whereas incubation of wildtype cardiomyocytes in high-glucose medium did not result in increased ROS generation. These results not only showed that hyperglycemia drives mitochondrial ROS production but also imply that certain diabetes-induced mitochondrial changes may predispose cardiac mitochondria of OVE26 mice to generate ROS [64]. Given that ETC defects can increase ROS generation by increasing the reductive state of the ETC, resulting in electron leakage, preexisting defects of the ETC could be one such predisposition for ROS production. Such defects in the ETC may result from direct protein damage, as has been exemplarily been shown for lipid peroxidation or protein tyrosine nitration of ETC complexes in STZ-diabetic rats [6066]. Insulin treatment removed lipid peroxidation of complex II and normalized respiration rates and complex II activity, suggesting ROS-induced protein damage in the ETC as a mechanism of mitochondrial dysfunction in these diabetic hearts. Similar to hyperglycemia, increased FAO may also increase ROS by increased electron delivery, but also by shifting fatty acyl-CoAs into pathways that mediate lipotoxicity, or induction of mitochondrial fission [455667].
- Furthermore, db/db cardiomyocytes simultaneously subjected to energetic stress and redox stress by hyperglycemia and isoproterenol treatment resulted in an impaired transition from state 4 to state 3 respiration, which accounted for increased ROS generation both from forward and RET [68]. This was likely related to decreased levels of glutathione (GSH) and Trx2, and the resulting more oxidizing environment correlated with impaired excitation-contraction coupling. Resetting the mitochondrial redox balance by exogenous application of GSH or with palmitate, mediated by the TrxR2/Trx2/Prx3 system, normalized excitation-contraction coupling, which led the authors to propose that the inability of the diabetic heart to deal with an increase in energy demand may result from a perturbation of the mitochondrial redox status [68]. Another mechanism which may support impaired function of antioxidant enzymes is reduced levels of the mitochondrial deacetylase SIRT3, which promotes increased acetylation and thereby inactivation of MnSOD [69]. Other recently discovered mechanisms favoring mitochondrial ROS production and promoting DbCM might be increased levels of mitochondrial calpains or MAOs [6570]. Mimicking the increased mitochondrial levels of calpain 1 in diabetic hearts by targeted upregulation of calpain-1 in mitochondria induced cleavage of the ATP5A1 subunit of the FOF1-ATPase, and increased O2·− production and apoptosis in cardiomyocytes. Conversely, selective inhibition of mitochondrial calpains attenuated disruption of ATP synthase, decreased mitochondrial O2·− production, and prevented apoptosis in cardiomyocytes of STZ-diabetic mice, suggesting increased mitochondrial calpains as another mitochondrial mechanism contributing to DbCM [65]. Levels of MAO-A and MAO-B were also increased in the same model of DM, and increased H2O2 production was markedly attenuated by administration of MAO inhibitors [70].
- Remodeling of the mitochondrial proteome
- Transcriptional regulation is generally considered the mode of choice to adapt to chronic stimuli or diseases, and impaired expression of ETC subunits has been proposed as a cause of impaired mitochondrial function in numerous diseases, including cardiac pathologies. In failing hearts, a concerted downregulation of ETC subunits has been proposed to contribute to impaired mitochondrial function and energy depletion [56]. In the diabetic heart, a number of ETC subunits have been reported to be downregulated using immunoblotting of single proteins, although inconsistent results have been observed between studies and models [15]. More details were revealed by recent proteomics analyses of heart mitochondria, when even the proteome of the two distinct mitochondrial subpopulations has been analyzed. Subsarcolemmal mitochondria from db/db hearts showed several impairments, including a decrease in size and internal complexity, displayed decreased state 3 respiration and ATP synthesis rates, decreased ETC complex activities, and increased oxidative damage, whereas interfibrillar mitochondria were literally unaffected. Using isobaric tags for relative and absolute quantification (iTRAQ) and multi-dimensional protein identification technologies, a predominant decrease of protein subunits of the ETC, the ATP synthase, and of proteins of the mitochondrial protein import machinery was revealed, whereas these protein were nearly unaffected in interfibrillar mitochondria [71]. While such remodeling of the ETC may impair electron flow through the ETC and thereby overall ATP regeneration, such ETC defects are also assumed to increase the reduced state of the ETC, thereby facilitating electron leak and subsequent generation of superoxide.
- Posttranslational modifications
- Chronically increased protein O-linked beta-N-acetylglucosamine glycosylation (O-GlcNAcylation) induced by hyperglycemia contributes to cardiomyocyte dysfunction in DM. Several protein targets of O-GlcNAcylation have been identified in the diabetic heart, including phospholamban, calmodulin-dependent protein kinase II (CAMKII), actin, GATA binding protein 4 (GATA4), cytochrome c oxidase subunit 1 (COX1), dynamin-related protein 1 (DRP1), or forkhead box protein O1 (FOXO1), indicating a potential impact on various pathways whose impairment have been proposed to contribute to DbCM (for review see [72]). It has been shown that hyperglycemia leads to increased O-GlcNAcylation of CaMKII and promotes CaMKII-dependent Ca2+ release from the sarcoplasmic reticulum (SR), while removal of O-GlcNAcylation from myofilaments of cardiomyocytes in a mouse model of T1DM could restore Ca2+ sensitivity [7374]. O-GlcNAcylation might also facilitate hypertrophic signaling, partially transmitted by the activation of different transcription factors such as nuclear factor of activated T-cells (NFAT), GATA4, or myocyte enhancer factor 2C (MEF2C) [7576]. With regards to mitochondrial mechanisms, O-GlcNAcomic profiling found that over 88 mitochondrial proteins can be O-GlcNAcylated during inhibition of O-GlcNAcase, with the OXPHOS system as a major target. This O-GlcNAcylation was associated with increased mitochondrial oxygen consumption rates, ATP production rates, and an enhanced threshold for mitochondrial permeability transition pore (mPTP) opening, indicating that O-GlcNAcylation regulates mitochondrial biology [77]. In neonatal rat cardiomyocytes incubated in hyperglycemic medium, several subunits of the ETC, such as NDUFA9 of complex I, subunits core 1 and core 2 of complex III, and subunit I of complex IV have been found to be O-GlcNAcylated [78]. This O-GlcNAcylation was however associated with impaired activity of complex I, III, and IV, and removal of O-GlcNAcylation by overexpression of O-GlcNAcase normalized complex activities, besides a normalization of ATP levels. In another study though, a mislocalization of O-GlcNAc transferase (OGT) within mitochondria, leading to impaired interaction of OGT with complex IV, has been proposed to be responsible for impaired complex IV activity in diabetic hearts [79]. Thus, O-GlcNAcylation represents an important posttranslational modification that regulates mitochondrial function, although the functional consequences of increased O-GlcNAcylation in diabetic hearts remains to be elucidated in more detail.
- Other modulators of posttranslational modifications within mitochondria are the protein family of SIRTs. SIRTs are nicotinamide adenine dinucleotide (NAD)+-dependent deacylases that are capable of removing a variety of different posttranslational modifications from protein lysine residues of a target protein, thereby regulating target protein function. SIRT3 is primarily localized within mitochondria and predominantly acts as a deacetylase, thereby counterbalancing the protein acetylation driven by acetyltransferases and non-enzymatic acetylation. Accordingly, lack of SIRT3 shifts the balance of acetylation towards increased protein acetylation, as was similarly observed in db/db hearts, including validated targets of SIRT3 such as LCAD and MnSOD [8081]. This increased protein acetylation in db/db hearts was associated with a decreased NAD+/NADH ratio and decreased expression and activity of SIRT3 [80]. Treatment with garlic or exogenous application of H2S restored the decreased NAD+/NADH ratio in db/db hearts and enhanced expression and activity of SIRT3, along with attenuation of hyperacetylation of ETC subunits and improvement of mitochondrial respiration and ATP synthesis [81]. In addition, H2S treatment normalized increased FAO rates and decreased pyruvate dehydrogenase (PDH) activity in db/db hearts, which improved respiratory function, ATP synthesis and ejection fraction, implying impaired SIRT3 activity in the pathogenesis of altered substrate utilization and impaired energetics in diabetic hearts. Accordingly, the authors proposed a model in which H2S administration improves the NAD+/NADH ratio and thus SIRT3 activity, leading to a myocardial substrate oxidation pattern that more closely resembles the physiologic myocardial substrate preference, thereby increasing the efficiency of ATP regeneration [80]. If true, it will be of interest to understand how cardiomyocytes will deal with the increased delivery of lipids within the cardiomyocytes in this setting, or if this normalization of the substrate oxidation pattern will in fact inhibit sarcolemmal FA uptake. In another study, cardiomyocyte apoptosis was increased and mitochondrial morphology (swelling, structural damage) was impaired in hearts of STZ-diabetic mice, accompanied by a suppression of autophagy, including mitophagy [82]. Prevention of a decrease in SIRT3 expression in neonatal mouse cardiomyocytes incubated in high glucose medium by SIRT3 overexpression restored autophagy and mitophagy and attenuated mitochondrial damage and apoptosis. In addition, overexpression of SIRT3 increased Parkin expression, suggesting that a suppression of SIRT3-Parkin signaling may have mediated the downregulation of mitophagy in diabetic hearts [82]. The potential role of the other mitochondrial SIRTs, SIRT4 and SIRT5, in the pathogenesis of DbCM and related mitochondrial defects remains to be elucidated.
- Mitophagy and mitochondrial biogenesis
- Autophagy represents a cellular mechanism by which damaged cellular components, including proteins, lipids, or cell organelles, are delivered to autophagosomes to ultimately degrade them following fusion with a lysosome. Autophagy has been shown to be crucial for the development, maturation and the function of the heart [83]. The specific targeting and removal of mitochondria by autophagy, i.e., mitophagy, is achieved by the pathways of phosphatase and tensin homolog-induced putative kinase 1 (PINK1) and the E3 ubiquitin ligase Parkin, and by many other proteins in the mitochondrial membrane or the cytosol [848586878889]. Recently, Tong et al. [90] showed that suppression of mitophagy either by deletion of autophagy-related protein kinase 7 (Atg7) or Parkin exacerbated DbCM in high-fat fed mice. Restoring mitophagy by injection of Tat-Beclin1 attenuated mitochondrial dysfunction, decreased lipid accumulation and protected against diastolic dysfunction [90]. In this study, high-fat diet upregulated mitophagy which highlights mitophagy as a protective compensatory mechanism in DbCM by which dysfunctional mitochondria can be dismissed. Together with the earlier made observations that PINK and Parkin levels are reduced in hearts of diabetic mice, one can speculate that either mitophagy is impaired in later stages of DM, or that the induction of mitophagy may not be sufficient to balance the mitochondrial damage caused by other mechanisms of DbCM [9192]. Another hypothesis would be that upregulated mitophagy is not adequately matched with subsequent mitochondrial biogenesis in diabetic hearts, thereby resulting in a decrease of total mitochondrial content.
- Removal of mitochondria by mitophagy needs to be counterbalanced by mitochondrial biogenesis to maintain mitochondrial content of the cell. Mitochondrial biogenesis is thus part of the physiological turnover process of mitochondria that is predominantly regulated by PCG-1α signaling, which coactivates multiple transcription factors such as PPARα, estrogen receptor-related α (ERRα), nuclear respiratory factor 1 and 2 (NRF1/2), and mitochondrial transcriptionfactor A (mtTFA) [9394]. In patients with T2DM, mitochondrial biogenesis may be reduced in different organs, including the heart [95969798]. In contrast, myocardial PGC-1α expression is rather increased in animal models of DM, along with increased mitochondrial DNA content and increased mitochondrial area as well as count in the hearts of diabetic mice, both in models of T1DM and T2DM [3499100101102]. Shen et al. [64] showed that, in OVE26 mice, increased mitochondrial area and number were accompanied by increased mitochondrial damage and reduced respiratory control ratio of the mitochondria. These observations may imply that, in rodent models of DbCM, increased mitochondrial biogenesis may be the attempt to generate new and functionally intact mitochondria to compensate for mitochondrial damage and dysfunction in diabetic hearts. However, in combination with impaired mitophagy, this process of increased mitochondrial biogenesis may not be sufficient to remove all damaged mitochondria and/or to replace them by newly generated intact mitochondria. Also, the possibility exists that newly generated mitochondria are immediately damaged again by persistent factors that induce mitochondrial damage (e.g., oxidative stress), or that the process of actual biogenesis of mitochondria per se may be defective, resulting in generation of rather defective than functionally intact mitochondria. Further studies are needed to clarify the complex role of mitochondrial biogenesis in the diabetic heart, to address the question why mitochondrial biogenesis and/or the respective signaling may be different between human and rodent DbCM, and whether therapeutic induction of mitochondrial biogenesis, as is discussed for many other cardiac pathologies, may represent a way to cure mitochondrial dysfunction in DbCM [103].
- Mitochondrial fission and fusion
- Mitochondria are dynamic organelles that undergo continuous fusion and fission, a process that is required for mitochondrial biogenesis and that contributes to the regulation of mitochondrial energetics and ROS homeostasis. Mitochondrial dynamics are mediated by the action of a variety of distinct proteins, including the fission proteins Drp1 and fission 1 (Fis1), and the fusion proteins mitofusion 1 and 2 (Mfn1/2) and optic atrophy 1 (Opa1), among others. Mitochondrial fusion seems to overweigh during nutrient starvation or energy demanding states, leading to elongated tubular mitochondria, whereas fission promotes smaller fragmented mitochondria and seems to be induced by caloric excess [104]. Fission and fusion also have an impact on mitophagy where fission can separate damaged segments of mitochondria and induce mitophagy, whereas mitochondria appear to be degraded less likely following fusion [94105]. In the heart, deletion of either fusion or fission related proteins like Mfn1 and 2 or Drp 1 can lead to cardiomyopathy [106107]. With regards to DM, Yu et al. [108] showed that incubation of H9C2 rat cardiomyoblasts in high glucose medium induced a rapid mitochondrial fragmentation via Drp1 signaling which led to overproduction of ROS. In hearts of T1DM rodents though, fragmentation of mitochondria rather occurs as a response to chronic hyperglycemia. While mitochondrial morphology was unaltered after 3 weeks of hyperglycemia, electron microscopy revealed distorted vacuous mitochondria with decreased matrix electron density after 5 weeks of hyperglycemia, accompanied by increased proteolytic cleavage of Opa1 leading to more fragmented mitochondria due to impaired fusion [109]. Of interest, ROS production was unaffected after 3 weeks of hyperglycemia, whereas after 5 weeks following hyperglycemia, dysfunction of the ETC complexes and increased ROS production (primarily from RET) were apparent in these hearts [109]. These observations rather suggest a progressive impairment in mitochondrial dynamics in DbCM that may be caused by a chronic dysbalance of fusion and fission. In addition, given that mitochondrial fission can increase ROS production and vice versa, a vicious cycle between ROS and mitochondrial fragmentation may develop during chronic hyperglycemia, with one mechanism being able to trigger and further impair the other. Support for a dysregulated balance of fusion and fission has also been demonstrated in a study of Makino et al. [110] who showed decreased levels of Opa1 in mouse coronary endothelial cells isolated from diabetic mice while Drp1 levels were also increased. Of further interest, lowering of oxidative stress by TEMPOL treatment restored normal mitochondrial morphology, suggesting that a chronic increase in ROS may trigger mitochondrial fragmentation in DbCM, and that ROS scavenging may be an effective way to interrupt the vicious cycle of increased ROS and impaired mitochondrial dynamics.
- Besides changes in expression, posttranslational modifications such as increased O-GlcNAcylation of Opa1 and Drp1 have also been shown to contribute to the changes in mitochondrial dynamics in the diabetic milieu. High glucose levels increase O-GlcNAcylation of Opa1, and reducing this modification attenuates mitochondrial dysfunction [111]. O-GlcNAcylation of Drp1 is observed in hearts of T2DM mice, which decreases phosphorylation of Drp1 and thereby induces translocation of Drp1 onto mitochondria and eventual fragmentation [112]. Insights into upstream mechanisms of impaired mitochondrial dynamics in DbCM come from observations in mice overexpressing long-chain acyl-CoA synthetase 1 (ACSL1) as a model of cardiac lipotoxicity. In this model, reduced phosphorylation of Drp1 and altered proteolytic processing of Opa1 led to increased mitochondrial fission, suggesting that increased lipid uptake, as typically observed in diabetic hearts, may trigger mitochondrial fragmentation and thus mitochondrial dysfunction and ROS [45]. The link between FA metabolism and mitochondrial dynamics was also endorsed by Kolleritsch et al. [113] who could show that cardiomyocyte-specific overexpression of mutated perilipin 5 resulted in reduced cardiac lipolysis and attenuation of mitochondrial fission, accompanied by less mitochondrial recruitment of Drp1 and decreased phosphorylation of the Drp1 interaction partner, mitochondrial fission factor. Of note, a recent study by Hu et al. [114] not only demonstrated that mitochondrial fission is impaired in T2DM db/db hearts, but also that the proposed mechanism of reduced Mfn2 expression may have been a consequence of reduced expression and binding of PPARα to the Mfn2 promoter, thus providing more evidence of a link between lipid metabolism and impaired mitochondrial dynamics in DbCM. According to these data, the authors proposed a model in which a decrease in PPARα expression in the rather chronic DM situation would impair expression of PPARα and Mfn2, thereby inducing mitochondrial fission which results in mitochondrial respiratory dysfunction, increased mitochondrial ROS generation and mitochondria-dependent apoptosis [114].
- Mitochondrial Ca2+ handling
- Ca2+ is the pivotal messenger for excitation-contraction coupling in the heart which connects the electrical stimulus to the contraction of the myocytes. During cardiomyocyte depolarization, a small Ca2+ influx via sarcolemmal L-type Ca2+ channels (LTCC) triggers opening of the ryanodine receptor (RyR) of the SR, resulting in a large release of Ca2+ from the SR into the cytosol, thereby activating myofilament cross-bridge formation and triggering cardiomyocyte contraction [115]. To end the contraction cycle, Ca2+ is mainly removed from the cytosol by reuptake into the SR via the sarco/ER Ca2+-ATPase 2a (SERCA2a), and a smaller amount is removed into the extracellular space via the Na+/Ca2+ exchanger [115]. To match increased energy demand with energy production, cytosolic Ca2+ transients also trigger mitochondrial Ca2+-uptake, facilitated by the mitochondrial calcium uniporter (MCU). In mitochondria, Ca2+ can stimulate ATP regeneration through enhancing the activity of ETC complexes I, II, and IV, and by activating PDH, α-ketoglutarate dehydrogenase, and isocitrate dehydrogenase. Furthermore, the FOF1-ATPase requires Ca2+ for its activity, and it has been estimated that this activation may have been responsible for more than 60% of the Ca2+-induced activation of OXPHOS, whereas the contribution of Ca2+-sensitive dehydrogenases may have accounted for only 40% [116].
- While there is evidence that cellular Ca2+ handling is impaired in DbCM due to decreased SERCA2a expression, reduced activity of RyR or decreased LTCC expression, literally no data on mitochondrial Ca2+ handling in DbCM have been published [117118]. However, studies showed that myocardial mitochondrial Ca2+ uptake was reduced in STZ-induced T1DM rats and in T2DM ob/ob mice [119120]. Recently, Ji et al. [121] found that cardiomyocyte expression of mitochondrial calcium uptake protein 1 (MICU1), a regulatory subunit of the MCU, was downregulated in db/db mice at 12 weeks of age, accompanied by mitochondria-dependent intrinsic apoptosis. Reconstitution of MICU1 normalized cardiac function, attenuated cardiac hypertrophy and fibrosis, and inhibited apoptosis in this mouse model [121]. In addition, increased mitochondrial Ca2+ uptake through upregulation of MICU1 attenuated mitochondrial ROS and ROS-triggered apoptosis [121]. In another study, expression of MICU1 was increased in hearts of STZ-diabetic mice, however levels of MCU and essential MCU regulator (EMRE; subunit of MCU) were decreased, thus also leading to a decrease in mitochondrial Ca2+ uptake, mitochondrial function, and cardiac function. Restoring MCU expression also rescued cardiac and mitochondrial respiratory dysfunction, underscoring the proposal of mitochondrial Ca2+ restoration as a potential target for therapeutic intervention [122123]. One has to keep in mind though that triggering of mitochondrial permeability transition due to Ca2+ overload may complicate this therapeutic strategy [124125]. Of note, increasing Ca2+ uptake by overexpression of MICU1 may be beneficial in diabetic hearts, but resulted in increased mortality in non-diabetic mice [121].
- Dysregulation of microRNAs
- MicroRNAs (miRNAs) are single-stranded non-coding RNA molecules that are about 22 nucleotides long and that regulate protein levels by inhibition of mRNA translation or degradation of mRNA [126]. Dysregulation of a large number of different miRNAs has been implicated by now to contribute to the pathogenesis of cardiovascular diseases, including HF and DbCM, as reviewed elsewhere [127128]. For example, miR133a expression was reduced in STZ-treated diabetic mice displaying ventricular dysfunction and hypertrophy, and transfection of miR133a mimics to cardiomyocytes incubated in high glucose medium prevented hypertrophic changes [129]. Also, miR-30c expression was decreased in diabetic mice with cardiomyocyte hypertrophy and in humans with DbCM, and overexpression of miR-30c attenuated hypertrophy in cardiomyocytes treated with glucose, possibly via decreased expression of cell division control protein 42 homolog (Cdc42) and p21activated kinase 1 (Pak1) [130]. Interestingly, it has been shown that not only nuclear-encoded proteins but also mitochondria-encoded proteins can be regulated by miRNAs [131132]. With regards to DbCM, a number of studies suggest that mitochondrial dysfunction may also result to some part from alterations in miRNA action. Overexpression of miRNA-195 observed in T1DM and T2DM hearts may contribute to downregulation of SIRT1 in these hearts, and impaired SIRT1 activity can then be assumed to downregulate oxidative metabolism, mitochondrial function and to increase ROS production [133]. PGC-1α is a direct target of miRNA-29a, and myocardial levels of miRNA-29a were shown to be decreased in STZ-induced diabetic animals, a mechanism that may contribute to induction of PPARα and FAO, as well as to mitochondrial biogenesis [134]. Baseler et al. [135] found increased levels of miRNA-141 in T1DM mice, which may impair the activity of solute carrier family 25 member 3 (Slc25a3) and thus import of inorganic phosphate and subsequent ATP regeneration. Also, an increased level of miRNA-378 in interfibrillar cardiac mitochondria of STZ-diabetic mice has been shown to impair the translation of the ATP6 subunit of the FOF1-ATPase [136]. These studies imply that miRNAs may interfere with different pathways, proteins and enzymes that are essential to maintain oxidative function of mitochondria. It is thus quite plausible that miRNA dysregulation contributes to mitochondrial dysfunction in DbCM, although the elucidation of exact contributions, the complexity of interactions, and the further characterization of all mitochondria-regulating miRNAs will be a major task of future studies.
MITOCHONDRIAL MECHANISMS OF DbCM
- Both our increased understanding of mitochondrial mechanisms contributing to DbCM and the increasing elucidation of mechanisms of action of antidiabetic drugs has increased the attention for mitochondria as a therapeutic target in diabetic heart disease. A number of antidiabetic medications currently used in clinical practice may already directly or indirectly attenuate mitochondrial defects associated with DbCM, including metformin. ER stress is a typical trait in DbCM, and a recent study demonstrated that induction of ER stress by thapsigargin treatment in non-diabetic mice impairs mitochondrial respiration, may induce mPTP opening, and increases mitochondrial oxidative stress [137]. Additional treatment of these mice with metformin normalized all of the mitochondrial abnormalities, potentially by activation of AMP activated protein kinase, suggesting that metformin may cure mitochondrial defects in DbCM induced by ER stress or even by other causes.
- Mostly discussed these days though are the potential mechanisms by which sodium glucose cotransporter 2 inhibitors (SGLT2i) improve cardiovascular outcomes in diabetic subjects. Results of several phase 3 clinical trials demonstrated that additional treatment of diabetic subjects with SGLT2i leads to a significant reduction in cardiovascular endpoints, including the classical major adverse cardiovascular event (MACE) endpoint (non-fatal myocardial infarction, non-fatal stroke, cardiovascular death), all-cause mortality, and/or in particular, hospitalization for HF [138139140]. Although each trial investigating outcomes of specific SGLT2i (empagliflozin, canagliflozin, dapagliflozin) showed slight differences in outcomes and patient inclusion criteria (patients with cardiovascular risk factors vs. patients with known cardiovascular disease), the effects of SGLT2i are generally considered rather a class effect. Accordingly, the European Society of Cardiology recently adapted the guidelines on the treatment of diabetic patients based on this new clinical trial evidence, now suggesting SGLT2i even as a first line therapy in diabetic subjects with high or very high cardiovascular risk or known cardiovascular disease [141]. Not included in the new guidelines yet are the results of the recent Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction (DAPA-HF) trial, which demonstrated that dapagliflozin treatment lead to a reduction of the composite endpoint of worsening HF or cardiovascular death in patients with HF with reduced ejection fraction, irrespective whether the patients suffered from diabetes or not [142]. Given the therapeutic efficacy of dapagliflozin in non-diabetic patients, the question rises in how far glucose lowering indeed contributes to the beneficial effects on macrovascular outcomes, mortality, and HF worsening. Furthermore, this trial indicates that SGLT2i treatment may target myocardial mechanisms underlying systolic HF per se, some of which are also present in patients with DbCM, including mitochondrial defects.
- Despite the likely absence of SGLT2 in the myocardium, direct myocardial mechanisms of SGLT2i have been identified or proposed that may affect mitochondrial function. The cardiac Na+/H+-exchanger 1 (NHE1) has been identified as a target of SGLT2i. In isolated cardiomyocytes from rabbits and mice, treatment with empagliflozin inhibited NHE1 flux, reduced cytosolic Na+ and Ca2+ levels, and increased mitochondrial Ca2+ levels, likely by direct binding of empagliflozin to NHE1 [143144]. SGLT2i may thereby attenuate defects in both cytosolic and mitochondrial Ca2+ handling and may increase ATP regeneration by activating mitochondrial Ca2+-sensitive dehydrogenases [143144]. Another mechanism may be related to SGLT2i-mediated alterations in myocardial ketone metabolism. SGLT2i treatment results in increased serum β-hydroxybutyrate levels which led to the “thrifty substrate” hypothesis, proposing that mild hyperketonemia and a subsequent relative increase in myocardial oxidation of ketone bodies may increase cardiac work per oxygen consumed, i.e., cardiac efficiency [144]. Indeed, continuous treatment with empagliflozin resulted in a reduction of glucose oxidation, in an increase in myocardial uptake of ketone bodies, FA, and branched-chain amino acids (BCAA), and in enhanced activity and/or expression of enzymes involved in ketone body, FA and BCAA metabolism in a porcine model of HF [145]. While this substrate switch was accompanied by increased systolic function and attenuation of left ventricular remodeling, it needs to be kept in mind that the animals were not diabetic and a similar effect needs to be demonstrated in diabetic animals as well [145]. Of note, subjecting pigs to myocardial ischemia reperfusion combined with short term pretreatment with canagliflozin preserved cardiac function and efficiency during ischemia but had no effect on the pattern of myocardial substrate uptake, including ketone bodies, potentially arguing against the “thrifty substrate” hypothesis, although energy metabolism during ischemia and reperfusion markedly differs from substrate utilization in chronic HF or DbCM [146].
- Similar to SGLT2i, some glucagon-like peptide 1 receptor agonists (GLP1RA) have demonstrated a reduction of cardiovascular endpoints in clinical trials, including MACE or CV death alone, thus resulting in a similar guideline recommendation as described before for SGLT2i [147148]. Although only few data are available on the underlying mechanisms of GLP1RA, mitochondrial mechanisms may also be affected by GLP1RA treatment. In a rat model of chronic hypoxia, treatment with liraglutide attenuated activation of the mitochondrial apoptosis pathway, increased activation of protective mitophagy via increased expression of Parkin, attenuated mitochondrial oxidative stress, and prevented energy depletion [149].
- Besides positive effects of antidiabetic drugs on mitochondrial biology in DbCM, other approaches may also be promising therapeutic strategies to improve mitochondrial function in DbCM. Given the multiple detrimental effects of mitochondrial oxidative stress in DbCM, mitochondrial ROS scavenging is an obvious potential treatment strategy in DbCM. Several mitochondria-targeted agents have demonstrated the capacity to attenuate mitochondrial oxidative stress in preclinical studies, such as MitoQ, MnTBAP, or MitoTempol. Treatment with MnTBAP reversed cardiac mitochondrial oxidative stress and improved mitochondrial bioenergetics in a mouse model of the metabolic syndrome [150]. In leukocytes of T2DM patients, treatment with MitoQ attenuated mitochondrial ROS production and showed antiinflammatory and antioxidant effects [151].While MitoQ has shown beneficial effects in different cardiac pathologies as reviewed elsewhere, studies evaluating antioxidative treatment in DbCM are however lacking [8152]. Another approach to attenuate mitochondrial oxidative damage is supplementation with tetrahydropterin (BH4), which is required for full functional activity of endothelial nitric oxidase synthase (eNOS). In diabetes, BH4 is oxidized to BH2, and low BH4 levels are known to result in decreased eNOS activity and in increased superoxide production by eNOS. Given the proximity of eNOS to the outer mitochondrial membrane, such eNOS-derived ROS may cause mitochondrial damage. Therapeutic application of sepiapterin, a BH4 precursor, together with L-citrulline as an L-arginine precursor, prevented cardiac dysfunction in diabetic db/db mice and also attenuated myocardial infarct size in this rodent model [153]. To date, none of the above discussed antioxidants have been investigated in human trials for potential beneficial effects in DbCM or HF.
- Some beneficial effect in DbCM may also be achieved by modulation of the myocardial (mitochondrial) substrate oxidation pattern towards a physiological substrate utilization pattern. For example, the anti-anginal agent trimetazidine inhibits long-chain 3-ketoacyl coenzyme A thiolase in the β-oxidation spiral and thereby reduces FAO, improves systolic function and exercise tolerance, and reduces N-terminal-pro hormone brain natriuretic peptide (NT-proBNP) levels in diabetic subjects with idiopathic dilated cardiomyopathy after 6 months of treatment when compared to placebo [154]. Furthermore, trimetazidine added to standard medical therapy showed beneficial effects on left ventricular ejection fraction in diabetic subjects with ischemic heart disease compared to placebo [155]. Ranolazine also inhibits FAO and activates PDH and has been shown to improve hemodynamics in HFpEF patients [156157]. In the Metabolic Efficiency with Ranolazine for Less Ischemia in Non-ST-Elevation Acute Coronary Syndromes trial (MERLIN-TIMI36) trial, ranolazine treatment reduced recurrent myocardial ischemia in diabetic patients [158]. Inhibition of CPT1 by perhexiline reduces FAO and has been shown to improve maximal oxygen uptake, to increase the phosphocreatine to creatine (PCr/Cr) ratio, improve left ventricular systolic function, as well as skeletal muscle energetics in chronic HF and cardiac energetics in patients with dilated cardiomyopathy [159160161162]. Data of ranolazine in patients with diabetic heart disease are lacking to date. Additional and larger trials investigating patients with DbCM with and without systolic HF are thus necessary to further evaluate a potential beneficial effect of metabolic modulation in diabetic subjects.
- Finally, NAD+ levels are depleted in DbCM, possibly due to hyperactivation of the NAD+ consuming DNA repair enzyme, poly (ADP-ribose)-polymerase 1 (PARP-1). Since NAD+ is an essential cosubstrate for SIRTs, cellular NAD+ depletion may impair the activity of both intramitochondrial and extramitochondrial SIRTs [43163]. A recently published study showed that chronic oral application of the NAD+ precursor nicotinamide riboside (NR) is well tolerated and able to elevate NAD+ levels in healthy adults, thereby opening a new treatment option for several cardiac diseases associated with decreased NAD+/NADH ratios, including DbCM [164]. In preclinical studies, activation of SIRTs by restoration of NAD+ levels by treatment with NAD precursors (nicotinamide mononucleotide or NR) or by overexpression of nicotinamide phosphoribosyltransferase improved cardiac function in different models of HF or following ischemia reperfusion [165166]. These effects have been proposed to be mediated by activation of SIRT1 and/or SIRT3. Pharmacological inhibition of PARP-1 would represent another approach to maintain cellular NAD+ levels. Indeed, application of the PARP-1 inhibitor INO1001 attenuated oxidative stress, inflammation, fibrosis, and increased expression of PGC-1α in hearts of T2DM mice via activation of SIRT1 [167].
POTENTIAL THERAPEUTIC STRATEGIES TARGETING MITOCHONDRIA IN DbCM
- In the current review, we discussed mechanisms and pathways that are disturbed in the diabetic heart and that render mitochondria susceptible to damage and dysfunction. These mitochondrial defects contribute to many pathologic features of the diabetic heart, including impaired contractility, which puts mitochondria at center stage of the pathophysiology of DbCM. The next step has to be a comprehensive confirmation of many of these mechanisms in the human diabetic heart to gain more insight if potential treatment targets identified in animal models may also be a valid target in humans. Such data are instrumental for the development of potential therapies to prevent or at least attenuate diabetes-related mitochondrial defects in the heart and to increase the chance that the therapeutic strategies also prove effective in clinical trials. The significance of improving myocardial mitochondrial abnormalities in diabetic subjects is emphasized by the fact that some beneficial effects of cardioprotective antidiabetic drugs (e.g., SGLT2i, metformin) seem to be mediated by direct or indirect effects on mitochondria. With increasing understanding of mitochondrial biology, the increasing use of large animal models, and by using rapidly evolving new scientific technologies, mitochondrial medicine may become a realistic therapeutic option in the closer future. Given the widespread involvement of mitochondrial defects in human disease, even including the pathogenesis of DM itself, the successful development of mitochondrial therapies will likely benefit numerous patients suffering from many other diseases beyond DbCM as well.
CONCLUSIONS
-
Acknowledgements
- None.
ACKNOWLEDGMENTS
-
CONFLICTS OF INTEREST: No potential conflict of interest relevant to this article was reported.
NOTES
- 1. Rubler S, Dlugash J, Yuceoglu YZ, Kumral T, Branwood AW, Grishman A. New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am J Cardiol 1972;30:595-602. ArticlePubMed
- 2. Bugger H, Abel ED. Molecular mechanisms of diabetic cardiomyopathy. Diabetologia 2014;57:660-671. ArticlePubMedPMCPDF
- 3. Kannel WB, Hjortland M, Castelli WP. Role of diabetes in congestive heart failure: the Framingham study. Am J Cardiol 1974;34:29-34. ArticlePubMed
- 4. Sarma S, Mentz RJ, Kwasny MJ, Fought AJ, Huffman M, Subacius H, Nodari S, Konstam M, Swedberg K, Maggioni AP, Zannad F, Bonow RO, Gheorghiade M. EVEREST investigators. Association between diabetes mellitus and post-discharge outcomes in patients hospitalized with heart failure: findings from the EVEREST trial. Eur J Heart Fail 2013;15:194-202. ArticlePubMed
- 5. Kristensen SL, Mogensen UM, Jhund PS, Petrie MC, Preiss D, Win S, Kober L, McKelvie RS, Zile MR, Anand IS, Komajda M, Gottdiener JS, Carson PE, McMurray JJ. Clinical and echocardiographic characteristics and cardiovascular outcomes according to diabetes status in patients with heart failure and preserved ejection fraction: a report from the I-Preserve trial (irbesartan in heart failure with preserved ejection fraction). Circulation 2017;135:724-735. ArticlePubMed
- 6. Nichols GA, Gullion CM, Koro CE, Ephross SA, Brown JB. The incidence of congestive heart failure in type 2 diabetes: an update. Diabetes Care 2004;27:1879-1884. PubMed
- 7. Dei Cas A, Fonarow GC, Gheorghiade M, Butler J. Concomitant diabetes mellitus and heart failure. Curr Probl Cardiol 2015;40:7-43. ArticlePubMed
- 8. Kenny HC, Abel ED. Heart failure in type 2 diabetes mellitus. Circ Res 2019;124:121-141. ArticlePubMedPMC
- 9. Maack C, Lehrke M, Backs J, Heinzel FR, Hulot JS, Marx N, Paulus WJ, Rossignol P, Taegtmeyer H, Bauersachs J, Bayes-Genis A, Brutsaert D, Bugger H, Clarke K, Cosentino F, De Keulenaer G, Dei Cas A, Gonzalez A, Huelsmann M, Iaccarino G, Lunde IG, Lyon AR, Pollesello P, Rena G, Riksen NP, Rosano G, Staels B, van Laake LW, Wanner C, Farmakis D, Filippatos G, Ruschitzka F, Seferovic P, de Boer RA, Heymans S. Heart failure and diabetes: metabolic alterations and therapeutic interventions: a state-of-the-art review from the Translational Research Committee of the Heart Failure Association-European Society of Cardiology. Eur Heart J 2018;39:4243-4254. ArticlePubMedPMC
- 10. Seferovic PM, Paulus WJ. Clinical diabetic cardiomyopathy: a two-faced disease with restrictive and dilated phenotypes. Eur Heart J 2015;36:1718-1727. ArticlePubMed
- 11. Jia G, DeMarco VG, Sowers JR. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat Rev Endocrinol 2016;12:144-153. ArticlePubMedPDF
- 12. Jia G, Hill MA, Sowers JR. Diabetic cardiomyopathy: an update of mechanisms contributing to this clinical entity. Circ Res 2018;122:624-638. ArticlePubMedPMC
- 13. Morino K, Petersen KF, Shulman GI. Molecular mechanisms of insulin resistance in humans and their potential links with mitochondrial dysfunction. Diabetes 2006;55 Suppl 2:S9-S15. ArticlePubMedPDF
- 14. Kuo TH, Giacomelli F, Wiener J. Oxidative metabolism of Polytron versus Nagarse mitochondria in hearts of genetically diabetic mice. Biochim Biophys Acta 1985;806:9-15. ArticlePubMed
- 15. Bugger H, Abel ED. Rodent models of diabetic cardiomyopathy. Dis Model Mech 2009;2:454-466. ArticlePubMedPDF
- 16. Anderson EJ, Kypson AP, Rodriguez E, Anderson CA, Lehr EJ, Neufer PD. Substrate-specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart. J Am Coll Cardiol 2009;54:1891-1898. ArticlePubMedPMC
- 17. Montaigne D, Marechal X, Coisne A, Debry N, Modine T, Fayad G, Potelle C, El Arid JM, Mouton S, Sebti Y, Duez H, Preau S, Remy-Jouet I, Zerimech F, Koussa M, Richard V, Neviere R, Edme JL, Lefebvre P, Staels B. Myocardial contractile dysfunction is associated with impaired mitochondrial function and dynamics in type 2 diabetic but not in obese patients. Circulation 2014;130:554-564. ArticlePubMed
- 18. Croston TL, Thapa D, Holden AA, Tveter KJ, Lewis SE, Shepherd DL, Nichols CE, Long DM, Olfert IM, Jagannathan R, Hollander JM. Functional deficiencies of subsarcolemmal mitochondria in the type 2 diabetic human heart. Am J Physiol Heart Circ Physiol 2014;307:H54-H65. ArticlePubMedPMC
- 19. Barth E, Stammler G, Speiser B, Schaper J. Ultrastructural quantitation of mitochondria and myofilaments in cardiac muscle from 10 different animal species including man. J Mol Cell Cardiol 1992;24:669-681. ArticlePubMed
- 20. Wisneski JA, Gertz EW, Neese RA, Gruenke LD, Craig JC. Dual carbon-labeled isotope experiments using D-[6-14C] glucose and L-[1,2,3-13C3] lactate: a new approach for investigating human myocardial metabolism during ischemia. J Am Coll Cardiol 1985;5:1138-1146. ArticlePubMed
- 21. Gertz EW, Wisneski JA, Stanley WC, Neese RA. Myocardial substrate utilization during exercise in humans. Dual carbon-labeled carbohydrate isotope experiments. J Clin Invest 1988;82:2017-2025. ArticlePubMedPMC
- 22. Bertero E, Maack C. Metabolic remodelling in heart failure. Nat Rev Cardiol 2018;15:457-470. ArticlePubMedPDF
- 23. Opie LH. Heart physiology: from cell to circulation. 4th ed. Philadelphia: Lippincott Williams & Wilkins; 2004.
- 24. Buchanan J, Mazumder PK, Hu P, Chakrabarti G, Roberts MW, Yun UJ, Cooksey RC, Litwin SE, Abel ED. Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology 2005;146:5341-5349. ArticlePubMedPDF
- 25. Wang P, Lloyd SG, Zeng H, Bonen A, Chatham JC. Impact of altered substrate utilization on cardiac function in isolated hearts from Zucker diabetic fatty rats. Am J Physiol Heart Circ Physiol 2005;288:H2102-H2110. ArticlePubMed
- 26. Ohtake T, Yokoyama I, Watanabe T, Momose T, Serezawa T, Nishikawa J, Sasaki Y. Myocardial glucose metabolism in noninsulin-dependent diabetes mellitus patients evaluated by FDG-PET. J Nucl Med 1995;36:456-463. PubMed
- 27. Peterson LR, Herrero P, Schechtman KB, Racette SB, Waggoner AD, Kisrieva-Ware Z, Dence C, Klein S, Marsala J, Meyer T, Gropler RJ. Effect of obesity and insulin resistance on myocardial substrate metabolism and efficiency in young women. Circulation 2004;109:2191-2196. ArticlePubMed
- 28. Peterson LR, Saeed IM, McGill JB, Herrero P, Schechtman KB, Gunawardena R, Recklein CL, Coggan AR, DeMoss AJ, Dence CS, Gropler RJ. Sex and type 2 diabetes: obesity-independent effects on left ventricular substrate metabolism and relaxation in humans. Obesity (Silver Spring) 2012;20:802-810. ArticlePubMedPDF
- 29. Rijzewijk LJ, van der Meer RW, Lamb HJ, de Jong HW, Lubberink M, Romijn JA, Bax JJ, de Roos A, Twisk JW, Heine RJ, Lammertsma AA, Smit JW, Diamant M. Altered myocardial substrate metabolism and decreased diastolic function in nonischemic human diabetic cardiomyopathy: studies with cardiac positron emission tomography and magnetic resonance imaging. J Am Coll Cardiol 2009;54:1524-1532. PubMed
- 30. Barger PM, Kelly DP. PPAR signaling in the control of cardiac energy metabolism. Trends Cardiovasc Med 2000;10:238-245. ArticlePubMed
- 31. Finck BN, Lehman JJ, Leone TC, Welch MJ, Bennett MJ, Kovacs A, Han X, Gross RW, Kozak R, Lopaschuk GD, Kelly DP. The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. J Clin Invest 2002;109:121-130. ArticlePubMedPMC
- 32. Mjos OD. Effect of free fatty acids on myocardial function and oxygen consumption in intact dogs. J Clin Invest 1971;50:1386-1389. ArticlePubMedPMC
- 33. Boudina S, Sena S, Theobald H, Sheng X, Wright JJ, Hu XX, Aziz S, Johnson JI, Bugger H, Zaha VG, Abel ED. Mitochondrial energetics in the heart in obesity-related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes 2007;56:2457-2466. PubMed
- 34. Boudina S, Sena S, O'Neill BT, Tathireddy P, Young ME, Abel ED. Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation 2005;112:2686-2695. ArticlePubMed
- 35. Boudina S, Bugger H, Sena S, O'Neill BT, Zaha VG, Ilkun O, Wright JJ, Mazumder PK, Palfreyman E, Tidwell TJ, Theobald H, Khalimonchuk O, Wayment B, Sheng X, Rodnick KJ, Centini R, Chen D, Litwin SE, Weimer BE, Abel ED. Contribution of impaired myocardial insulin signaling to mitochondrial dysfunction and oxidative stress in the heart. Circulation 2009;119:1272-1283. ArticlePubMedPMC
- 36. Bugger H, Riehle C, Jaishy B, Wende AR, Tuinei J, Chen D, Soto J, Pires KM, Boudina S, Theobald HA, Luptak I, Wayment B, Wang X, Litwin SE, Weimer BC, Abel ED. Genetic loss of insulin receptors worsens cardiac efficiency in diabetes. J Mol Cell Cardiol 2012;52:1019-1026. ArticlePubMedPMC
- 37. Mazumder PK, O'Neill BT, Roberts MW, Buchanan J, Yun UJ, Cooksey RC, Boudina S, Abel ED. Impaired cardiac efficiency and increased fatty acid oxidation in insulin-resistant ob/ob mouse hearts. Diabetes 2004;53:2366-2374. ArticlePubMedPDF
- 38. Zlobine I, Gopal K, Ussher JR. Lipotoxicity in obesity and diabetes-related cardiac dysfunction. Biochim Biophys Acta 2016;1861:1555-1568. ArticlePubMed
- 39. Sharma S, Adrogue JV, Golfman L, Uray I, Lemm J, Youker K, Noon GP, Frazier OH, Taegtmeyer H. Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB J 2004;18:1692-1700. ArticlePubMedPDF
- 40. McGavock JM, Lingvay I, Zib I, Tillery T, Salas N, Unger R, Levine BD, Raskin P, Victor RG, Szczepaniak LS. Cardiac steatosis in diabetes mellitus: a 1H-magnetic resonance spectroscopy study. Circulation 2007;116:1170-1175. ArticlePubMed
- 41. Dong S, Zhang R, Liang Y, Shi J, Li J, Shang F, Mao X, Sun J. Changes of myocardial lipidomics profiling in a rat model of diabetic cardiomyopathy using UPLC/Q-TOF/MS analysis. Diabetol Metab Syndr 2017;9:56ArticlePubMedPMCPDF
- 42. Sletten AC, Peterson LR, Schaffer JE. Manifestations and mechanisms of myocardial lipotoxicity in obesity. J Intern Med 2018;284:478-491. ArticlePubMedPMCPDF
- 43. Gollmer J, Zirlik A, Bugger H. Established and emerging mechanisms of diabetic cardiomyopathy. J Lipid Atheroscler 2019;8:26-47.ArticlePubMedPMCPDF
- 44. Park TS, Hu Y, Noh HL, Drosatos K, Okajima K, Buchanan J, Tuinei J, Homma S, Jiang XC, Abel ED, Goldberg IJ. Ceramide is a cardiotoxin in lipotoxic cardiomyopathy. J Lipid Res 2008;49:2101-2112. ArticlePubMedPMC
- 45. Tsushima K, Bugger H, Wende AR, Soto J, Jenson GA, Tor AR, McGlauflin R, Kenny HC, Zhang Y, Souvenir R, Hu XX, Sloan CL, Pereira RO, Lira VA, Spitzer KW, Sharp TL, Shoghi KI, Sparagna GC, Rog-Zielinska EA, Kohl P, Khalimonchuk O, Schaffer JE, Abel ED. Mitochondrial reactive oxygen species in lipotoxic hearts induce post-translational modifications of AKAP121, DRP1, and OPA1 that promote mitochondrial fission. Circ Res 2018;122:58-73. ArticlePubMed
- 46. Chiu HC, Kovacs A, Ford DA, Hsu FF, Garcia R, Herrero P, Saffitz JE, Schaffer JE. A novel mouse model of lipotoxic cardiomyopathy. J Clin Invest 2001;107:813-822. ArticlePubMedPMC
- 47. Dodd MS, Sousa Fialho MDL, Montes Aparicio CN, Kerr M, Timm KN, Griffin JL, Luiken JJFP, Glatz JFC, Tyler DJ, Heather LC. Fatty acids prevent hypoxia-inducible factor-1α signaling through decreased succinate in diabetes. JACC Basic Transl Sci 2018;3:485-498. ArticlePubMedPMC
- 48. Shibata R, Murohara T, Ouchi N. Protective role of adiponectin in cardiovascular disease. Curr Med Chem 2012;19:5459-5466. ArticlePubMed
- 49. Yan W, Zhang H, Liu P, Wang H, Liu J, Gao C, Liu Y, Lian K, Yang L, Sun L, Guo Y, Zhang L, Dong L, Lau WB, Gao E, Gao F, Xiong L, Wang H, Qu Y, Tao L. Impaired mitochondrial biogenesis due to dysfunctional adiponectin-AMPK-PGC-1α signaling contributing to increased vulnerability in diabetic heart. Basic Res Cardiol 2013;108:329ArticlePubMedPDF
- 50. Civitarese AE, Ukropcova B, Carling S, Hulver M, DeFronzo RA, Mandarino L, Ravussin E, Smith SR. Role of adiponectin in human skeletal muscle bioenergetics. Cell Metab 2006;4:75-87. ArticlePubMedPMC
- 51. Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S, Yamashita S, Noda M, Kita S, Ueki K, Eto K, Akanuma Y, Froguel P, Foufelle F, Ferre P, Carling D, Kimura S, Nagai R, Kahn BB, Kadowaki T. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med 2002;8:1288-1295. ArticlePubMedPDF
- 52. Yamauchi T, Kamon J, Ito Y, Tsuchida A, Yokomizo T, Kita S, Sugiyama T, Miyagishi M, Hara K, Tsunoda M, Murakami K, Ohteki T, Uchida S, Takekawa S, Waki H, Tsuno NH, Shibata Y, Terauchi Y, Froguel P, Tobe K, Koyasu S, Taira K, Kitamura T, Shimizu T, Nagai R, Kadowaki T. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature 2003;423:762-769. ArticlePubMedPDF
- 53. Iwabu M, Yamauchi T, Okada-Iwabu M, Sato K, Nakagawa T, Funata M, Yamaguchi M, Namiki S, Nakayama R, Tabata M, Ogata H, Kubota N, Takamoto I, Hayashi YK, Yamauchi N, Waki H, Fukayama M, Nishino I, Tokuyama K, Ueki K, Oike Y, Ishii S, Hirose K, Shimizu T, Touhara K, Kadowaki T. Adiponectin and AdipoR1 regulate PGC-1alpha and mitochondria by Ca(2+) and AMPK/SIRT1. Nature 2010;464:1313-1319. ArticlePubMedPDF
- 54. Yamauchi T, Nio Y, Maki T, Kobayashi M, Takazawa T, Iwabu M, Okada-Iwabu M, Kawamoto S, Kubota N, Kubota T, Ito Y, Kamon J, Tsuchida A, Kumagai K, Kozono H, Hada Y, Ogata H, Tokuyama K, Tsunoda M, Ide T, Murakami K, Awazawa M, Takamoto I, Froguel P, Hara K, Tobe K, Nagai R, Ueki K, Kadowaki T. Targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and metabolic actions. Nat Med 2007;13:332-339. ArticlePubMedPDF
- 55. Koentges C, Konig A, Pfeil K, Holscher ME, Schnick T, Wende AR, Schrepper A, Cimolai MC, Kersting S, Hoffmann MM, Asal J, Osterholt M, Odening KE, Doenst T, Hein L, Abel ED, Bode C, Bugger H. Myocardial mitochondrial dysfunction in mice lacking adiponectin receptor 1. Basic Res Cardiol 2015;110:37. ArticlePubMedPDF
- 56. Bugger H, Abel ED. Mitochondria in the diabetic heart. Cardiovasc Res 2010;88:229-240. ArticlePubMedPMC
- 57. Murphy MP. How mitochondria produce reactive oxygen species. Biochem J 2009;417:1-13. ArticlePubMedPDF
- 58. Angelova PR, Abramov AY. Functional role of mitochondrial reactive oxygen species in physiology. Free Radic Biol Med 2016;100:81-85. ArticlePubMed
- 59. Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Mol Cell 2012;48:158-167. ArticlePubMedPMC
- 60. Lashin OM, Szweda PA, Szweda LI, Romani AM. Decreased complex II respiration and HNE-modified SDH subunit in diabetic heart. Free Radic Biol Med 2006;40:886-896. ArticlePubMed
- 61. Ye G, Metreveli NS, Donthi RV, Xia S, Xu M, Carlson EC, Epstein PN. Catalase protects cardiomyocyte function in models of type 1 and type 2 diabetes. Diabetes 2004;53:1336-1343. ArticlePubMedPDF
- 62. Cong W, Ruan D, Xuan Y, Niu C, Tao Y, Wang Y, Zhan K, Cai L, Jin L, Tan Y. Cardiac-specific overexpression of catalase prevents diabetes-induced pathological changes by inhibiting NF-κB signaling activation in the heart. J Mol Cell Cardiol 2015;89:314-325. ArticlePubMed
- 63. Arkat S, Umbarkar P, Singh S, Sitasawad SL. Mitochondrial peroxiredoxin-3 protects against hyperglycemia induced myocardial damage in diabetic cardiomyopathy. Free Radic Biol Med 2016;97:489-500. ArticlePubMed
- 64. Shen X, Zheng S, Metreveli NS, Epstein PN. Protection of cardiac mitochondria by overexpression of MnSOD reduces diabetic cardiomyopathy. Diabetes 2006;55:798-805. ArticlePubMedPDF
- 65. Ni R, Cao T, Xiong S, Ma J, Fan GC, Lacefield JC, Lu Y, Le Tissier S, Peng T. Therapeutic inhibition of mitochondrial reactive oxygen species with mito-TEMPO reduces diabetic cardiomyopathy. Free Radic Biol Med 2016;90:12-23. ArticlePubMed
- 66. Turko IV, Li L, Aulak KS, Stuehr DJ, Chang JY, Murad F. Protein tyrosine nitration in the mitochondria from diabetic mouse heart. Implications to dysfunctional mitochondria in diabetes. J Biol Chem 2003;278:33972-33977. PubMed
- 67. Korshunov SS, Skulachev VP, Starkov AA. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett 1997;416:15-18. ArticlePubMedPDF
- 68. Tocchetti CG, Caceres V, Stanley BA, Xie C, Shi S, Watson WH, O'Rourke B, Spadari-Bratfisch RC, Cortassa S, Akar FG, Paolocci N, Aon MA. GSH or palmitate preserves mitochondrial energetic/redox balance, preventing mechanical dysfunction in metabolically challenged myocytes/hearts from type 2 diabetic mice. Diabetes 2012;61:3094-3105. ArticlePubMedPMCPDF
- 69. Sultana MR, Bagul PK, Katare PB, Anwar Mohammed S, Padiya R, Banerjee SK. Garlic activates SIRT-3 to prevent cardiac oxidative stress and mitochondrial dysfunction in diabetes. Life Sci 2016;164:42-51. ArticlePubMed
- 70. Sturza A, Duicu OM, Vaduva A, Danila MD, Noveanu L, Varro A, Muntean DM. Monoamine oxidases are novel sources of cardiovascular oxidative stress in experimental diabetes. Can J Physiol Pharmacol 2015;93:555-561. ArticlePubMed
- 71. Dabkowski ER, Baseler WA, Williamson CL, Powell M, Razunguzwa TT, Frisbee JC, Hollander JM. Mitochondrial dysfunction in the type 2 diabetic heart is associated with alterations in spatially distinct mitochondrial proteomes. Am J Physiol Heart Circ Physiol 2010;299:H529-H540. ArticlePubMedPMC
- 72. Ducheix S, Magre J, Cariou B, Prieur X. Chronic O-GlcNAcylation and diabetic cardiomyopathy: the bitterness of glucose. Front Endocrinol (Lausanne) 2018;9:642. ArticlePubMedPMC
- 73. Erickson JR, Pereira L, Wang L, Han G, Ferguson A, Dao K, Copeland RJ, Despa F, Hart GW, Ripplinger CM, Bers DM. Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature 2013;502:372-376. ArticlePubMedPMCPDF
- 74. Ramirez-Correa GA, Ma J, Slawson C, Zeidan Q, Lugo-Fagundo NS, Xu M, Shen X, Gao WD, Caceres V, Chakir K, DeVine L, Cole RN, Marchionni L, Paolocci N, Hart GW, Murphy AM. Removal of abnormal myofilament O-GlcNAcylation restores Ca2+ sensitivity in diabetic cardiac muscle. Diabetes 2015;64:3573-3587. ArticlePubMedPMCPDF
- 75. Facundo HT, Brainard RE, Watson LJ, Ngoh GA, Hamid T, Prabhu SD, Jones SP. O-GlcNAc signaling is essential for NFAT-mediated transcriptional reprogramming during cardiomyocyte hypertrophy. Am J Physiol Heart Circ Physiol 2012;302:H2122-H2130. ArticlePubMedPMC
- 76. Cannon MV, Sillje HH, Sijbesma JW, Vreeswijk-Baudoin I, Ciapaite J, van der Sluis B, van Deursen J, Silva GJ, de Windt LJ, Gustafsson JA, van der Harst P, van Gilst WH, de Boer RA. Cardiac LXRα protects against pathological cardiac hypertrophy and dysfunction by enhancing glucose uptake and utilization. EMBO Mol Med 2015;7:1229-1243. ArticlePubMedPMCPDF
- 77. Ma J, Liu T, Wei AC, Banerjee P, O'Rourke B, Hart GW. O-GlcNAcomic profiling identifies widespread O-linked β-N-acetylglucosamine modification (O-GlcNAcylation) in oxidative phosphorylation system regulating cardiac mitochondrial function. J Biol Chem 2015;290:29141-29153. ArticlePubMedPMC
- 78. Hu Y, Suarez J, Fricovsky E, Wang H, Scott BT, Trauger SA, Han W, Hu Y, Oyeleye MO, Dillmann WH. Increased enzymatic O-GlcNAcylation of mitochondrial proteins impairs mitochondrial function in cardiac myocytes exposed to high glucose. J Biol Chem 2009;284:547-555. ArticlePubMedPMC
- 79. Banerjee PS, Ma J, Hart GW. Diabetes-associated dysregulation of O-GlcNAcylation in rat cardiac mitochondria. Proc Natl Acad Sci U S A 2015;112:6050-6055. ArticlePubMedPMC
- 80. Sun Y, Tian Z, Liu N, Zhang L, Gao Z, Sun X, Yu M, Wu J, Yang F, Zhao Y, Ren H, Chen H, Zhao D, Wang Y, Dong S, Xu C, Lu F, Zhang W. Exogenous H(2)S switches cardiac energy substrate metabolism by regulating SIRT3 expression in db/db mice. J Mol Med (Berl) 2018;96:281-299. ArticlePubMedPDF
- 81. Sun Y, Teng Z, Sun X, Zhang L, Chen J, Wang B, Lu F, Liu N, Yu M, Peng S, Wang Y, Zhao D, Zhao Y, Ren H, Cheng Z, Dong S, Lu F, Zhang W. Exogenous H(2)S reduces the acetylation levels of mitochondrial respiratory enzymes via regulating the NAD(+)-SIRT3 pathway in cardiac tissues of db/db mice. Am J Physiol Endocrinol Metab 2019;317:E284-E297. ArticlePubMedPMC
- 82. Yu W, Gao B, Li N, Wang J, Qiu C, Zhang G, Liu M, Zhang R, Li C, Ji G, Zhang Y. Sirt3 deficiency exacerbates diabetic cardiac dysfunction: role of Foxo3A-Parkin-mediated mitophagy. Biochim Biophys Acta Mol Basis Dis 2017;1863:1973-1983. ArticlePubMed
- 83. Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, Nishida K, Hori M, Mizushima N, Otsu K. The role of autophagy in in the basal state and in response to hemodynamic stress. Nat Med 2007;13:619-624. ArticlePubMedPDF
- 84. Ding WX, Ni HM, Li M, Liao Y, Chen X, Stolz DB, Dorn GW 2nd, Yin XM. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J Biol Chem 2010;285:27879-27890. ArticlePubMedPMC
- 85. Zhang J, Ney PA. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ 2009;16:939-946. ArticlePubMedPDF
- 86. Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, Ma Q, Zhu C, Wang R, Qi W, Huang L, Xue P, Li B, Wang X, Jin H, Wang J, Yang F, Liu P, Zhu Y, Sui S, Chen Q. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol 2012;14:177-185. ArticlePubMedPDF
- 87. Murakawa Y, Hinz M, Mothes J, Schuetz A, Uhl M, Wyler E, Yasuda T, Mastrobuoni G, Friedel CC, Dolken L, Kempa S, Schmidt-Supprian M, Bluthgen N, Backofen R, Heinemann U, Wolf J, Scheidereit C, Landthaler M. RC3H1 post-transcriptionally regulates A20 mRNA and modulates the activity of the IKK/NF-κB pathway. Nat Commun 2015;6:7367. ArticlePubMedPDF
- 88. Otsu K, Murakawa T, Yamaguchi O. BCL2L13 is a mammalian homolog of the yeast mitophagy receptor Atg32. Autophagy 2015;11:1932-1933. ArticlePubMedPMC
- 89. Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015;524:309-314. ArticlePubMedPMCPDF
- 90. Tong M, Saito T, Zhai P, Oka SI, Mizushima W, Nakamura M, Ikeda S, Shirakabe A, Sadoshima J. Mitophagy is essential for maintaining cardiac function during high fat diet-induced diabetic cardiomyopathy. Circ Res 2019;124:1360-1371. ArticlePubMedPMC
- 91. Xu X, Kobayashi S, Chen K, Timm D, Volden P, Huang Y, Gulick J, Yue Z, Robbins J, Epstein PN, Liang Q. Diminished autophagy limits cardiac injury in mouse models of type 1 diabetes. J Biol Chem 2013;288:18077-18092. ArticlePubMedPMC
- 92. Tang Y, Liu J, Long J. Phosphatase and tensin homolog-induced putative kinase 1 and Parkin in diabetic heart: role of mitophagy. J Diabetes Investig 2015;6:250-255.ArticlePubMedPDF
- 93. Fernandez-Marcos PJ, Auwerx J. Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. Am J Clin Nutr 2011;93:884S-8890. ArticlePubMedPMC
- 94. Liang Q, Kobayashi S. Mitochondrial quality control in the diabetic heart. J Mol Cell Cardiol 2016;95:57-69. ArticlePubMed
- 95. Karamanlidis G, Nascimben L, Couper GS, Shekar PS, del Monte F, Tian R. Defective DNA replication impairs mitochondrial biogenesis in human failing hearts. Circ Res 2010;106:1541-1548. ArticlePubMedPMC
- 96. Ritov VB, Menshikova EV, He J, Ferrell RE, Goodpaster BH, Kelley DE. Deficiency of subsarcolemmal mitochondria in obesity and type 2 diabetes. Diabetes 2005;54:8-14. ArticlePubMedPDF
- 97. Nisoli E, Clementi E, Carruba MO, Moncada S. Defective mitochondrial biogenesis: a hallmark of the high cardiovascular risk in the metabolic syndrome? Circ Res 2007;100:795-806. PubMed
- 98. Ren J, Pulakat L, Whaley-Connell A, Sowers JR. Mitochondrial biogenesis in the metabolic syndrome and cardiovascular disease. J Mol Med (Berl) 2010;88:993-1001. ArticlePubMedPMCPDF
- 99. Shen X, Ye G, Metreveli NS, Epstein PN. Cardiomyocyte defects in diabetic models and protection with cardiac-targeted transgenes. Methods Mol Med 2005;112:379-388. ArticlePubMed
- 100. Boudina S, Abel ED. Mitochondrial uncoupling: a key contributor to reduced cardiac efficiency in diabetes. Physiology (Bethesda) 2006;21:250-258. ArticlePubMed
- 101. Bugger H, Abel ED. Molecular mechanisms for myocardial mitochondrial dysfunction in the metabolic syndrome. Clin Sci (Lond) 2008;114:195-210. ArticlePubMedPDF
- 102. Duncan JG, Fong JL, Medeiros DM, Finck BN, Kelly DP. Insulin-resistant heart exhibits a mitochondrial biogenic response driven by the peroxisome proliferator-activated receptor-alpha/ PGC-1alpha gene regulatory pathway. Circulation 2007;115:909-917. ArticlePubMedPMC
- 103. Schilling J, Kelly DP. The PGC-1 cascade as a therapeutic target for heart failure. J Mol Cell Cardiol 2011;51:578-583. ArticlePubMed
- 104. Abel ED. Mitochondrial dynamics and metabolic regulation in cardiac and skeletal muscle. Trans Am Clin Climatol Assoc 2018;129:266-278. PubMedPMC
- 105. Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol 2011;13:589-598. ArticlePubMedPMCPDF
- 106. Chen Y, Liu Y, Dorn GW 2nd. Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ Res 2011;109:1327-1331. ArticlePubMedPMC
- 107. Ikeda Y, Shirakabe A, Maejima Y, Zhai P, Sciarretta S, Toli J, Nomura M, Mihara K, Egashira K, Ohishi M, Abdellatif M, Sadoshima J. Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ Res 2015;116:264-278. ArticlePubMed
- 108. Yu T, Robotham JL, Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc Natl Acad Sci U S A 2006;103:2653-2658. ArticlePubMedPMC
- 109. Galloway CA, Yoon Y. Mitochondrial dynamics in diabetic cardiomyopathy. Antioxid Redox Signal 2015;22:1545-1562. ArticlePubMedPMC
- 110. Makino A, Scott BT, Dillmann WH. Mitochondrial fragmentation and superoxide anion production in coronary endothelial cells from a mouse model of type 1 diabetes. Diabetologia 2010;53:1783-1794. ArticlePubMedPMC
- 111. Makino A, Suarez J, Gawlowski T, Han W, Wang H, Scott BT, Dillmann WH. Regulation of mitochondrial morphology and function by O-GlcNAcylation in neonatal cardiac myocytes. Am J Physiol Regul Integr Comp Physiol 2011;300:R1296-R1302. ArticlePubMedPMC
- 112. Gawlowski T, Suarez J, Scott B, Torres-Gonzalez M, Wang H, Schwappacher R, Han X, Yates JR 3rd, Hoshijima M, Dillmann W. Modulation of dynamin-related protein 1 (DRP1) function by increased O-linked-β-N-acetylglucosamine modification (O-GlcNAc) in cardiac myocytes. J Biol Chem 2012;287:30024-30034. ArticlePubMedPMC
- 113. Kolleritsch S, Kien B, Schoiswohl G, Diwoky C, Schreiber R, Heier C, Maresch LK, Schweiger M, Eichmann TO, Stryeck S, Krenn P, Tomin T, Kolb D, Rulicke T, Hoefler G, Wolinski H, Madl T, Birner-Gruenberger R, Haemmerle G. Low cardiac lipolysis reduces mitochondrial fission and prevents lipotoxic heart dysfunction in perilipin 5 mutant mice. Cardiovasc Res 2020;116:339-352. ArticlePubMedPDF
- 114. Hu L, Ding M, Tang D, Gao E, Li C, Wang K, Qi B, Qiu J, Zhao H, Chang P, Fu F, Li Y. Targeting mitochondrial dynamics by regulating Mfn2 for therapeutic intervention in diabetic cardiomyopathy. Theranostics 2019;9:3687-3706. ArticlePubMedPMC
- 115. Bertero E, Maack C. Reply to ‘Metabolic remodelling in heart failure revisited’. Nat Rev Cardiol 2018;15:780-781.ArticlePDF
- 116. Territo PR, Mootha VK, French SA, Balaban RS. Ca(2+) activation of heart mitochondrial oxidative phosphorylation: role of the F(0)/F(1)-ATPase. Am J Physiol Cell Physiol 2000;278:C423-C435. ArticlePubMed
- 117. Pereira L, Matthes J, Schuster I, Valdivia HH, Herzig S, Richard S, Gomez AM. Mechanisms of [Ca2+]i transient decrease in cardiomyopathy of db/db type 2 diabetic mice. Diabetes 2006;55:608-615. ArticlePubMedPDF
- 118. Pereira L, Ruiz-Hurtado G, Rueda A, Mercadier JJ, Benitah JP, Gomez AM. Calcium signaling in diabetic cardiomyocytes. Cell Calcium 2014;56:372-380. ArticlePubMed
- 119. Oliveira PJ, Seica R, Coxito PM, Rolo AP, Palmeira CM, Santos MS, Moreno AJ. Enhanced permeability transition explains the reduced calcium uptake in cardiac mitochondria from streptozotocin-induced diabetic rats. FEBS Lett 2003;554:511-514. ArticlePubMed
- 120. Fauconnier J, Lanner JT, Zhang SJ, Tavi P, Bruton JD, Katz A, Westerblad H. Insulin and inositol 1,4,5-trisphosphate trigger abnormal cytosolic Ca2+ transients and reveal mitochondrial Ca2+ handling defects in cardiomyocytes of ob/ob mice. Diabetes 2005;54:2375-2381. ArticlePubMedPDF
- 121. Ji L, Liu F, Jing Z, Huang Q, Zhao Y, Cao H, Li J, Yin C, Xing J, Li F. MICU1 alleviates diabetic cardiomyopathy through mitochondrial Ca(2+)-dependent antioxidant response. Diabetes 2017;66:1586-1600. ArticlePubMedPDF
- 122. Suarez J, Cividini F, Scott BT, Lehmann K, Diaz-Juarez J, Diemer T, Dai A, Suarez JA, Jain M, Dillmann WH. Restoring mitochondrial calcium uniporter expression in diabetic mouse heart improves mitochondrial calcium handling and cardiac function. J Biol Chem 2018;293:8182-8195. ArticlePubMedPMC
- 123. Dillmann WH. Diabetic cardiomyopathy. Circ Res 2019;124:1160-1162. ArticlePubMedPMC
- 124. Crompton M, Ellinger H, Costi A. Inhibition by cyclosporin A of a Ca2+-dependent pore in heart mitochondria activated by inorganic phosphate and oxidative stress. Biochem J 1988;255:357-360. PubMedPMC
- 125. Griffiths EJ, Halestrap AP. Further evidence that cyclosporin A protects mitochondria from calcium overload by inhibiting a matrix peptidyl-prolyl cis-trans isomerase. Implications for the immunosuppressive and toxic effects of cyclosporin. Biochem J 1991;274:611-614. ArticlePubMedPMCPDF
- 126. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell 2009;136:215-233. ArticlePubMedPMC
- 127. Hathaway QA, Pinti MV, Durr AJ, Waris S, Shepherd DL, Hollander JM. Regulating microRNA expression: at the heart of diabetes mellitus and the mitochondrion. Am J Physiol Heart Circ Physiol 2018;314:H293-H310. ArticlePubMed
- 128. Guo R, Nair S. Role of microRNA in diabetic cardiomyopathy: from mechanism to intervention. Biochim Biophys Acta Mol Basis Dis 2017;1863:2070-2077. ArticlePubMed
- 129. Feng B, Chen S, George B, Feng Q, Chakrabarti S. miR133a regulates cardiomyocyte hypertrophy in diabetes. Diabetes Metab Res Rev 2010;26:40-49. ArticlePubMed
- 130. Raut SK, Kumar A, Singh GB, Nahar U, Sharma V, Mittal A, Sharma R, Khullar M. miR-30c mediates upregulation of cdc42 and pak1 in diabetic cardiomyopathy. Cardiovasc Ther 2015;33:89-97. ArticlePubMed
- 131. Das S, Ferlito M, Kent OA, Fox-Talbot K, Wang R, Liu D, Raghavachari N, Yang Y, Wheelan SJ, Murphy E, Steenbergen C. Nuclear miRNA regulates the mitochondrial genome in the heart. Circ Res 2012;110:1596-1603. ArticlePubMedPMC
- 132. Baradan R, Hollander JM, Das S. Mitochondrial miRNAs in diabetes: just the tip of the iceberg. Can J Physiol Pharmacol 2017;95:1156-1162. ArticlePubMedPMC
- 133. Zheng D, Ma J, Yu Y, Li M, Ni R, Wang G, Chen R, Li J, Fan GC, Lacefield JC, Peng T. Silencing of miR-195 reduces diabetic cardiomyopathy in C57BL/6 mice. Diabetologia 2015;58:1949-1958. ArticlePubMedPMCPDF
- 134. Diao X, Shen E, Wang X, Hu B. Differentially expressed microRNAs and their target genes in the hearts of streptozotocin-induced diabetic mice. Mol Med Rep 2011;4:633-640. PubMed
- 135. Baseler WA, Thapa D, Jagannathan R, Dabkowski ER, Croston TL, Hollander JM. miR-141 as a regulator of the mitochondrial phosphate carrier (Slc25a3) in the type 1 diabetic heart. Am J Physiol Cell Physiol 2012;303:C1244-C1251. ArticlePubMedPMC
- 136. Jagannathan R, Thapa D, Nichols CE, Shepherd DL, Stricker JC, Croston TL, Baseler WA, Lewis SE, Martinez I, Hollander JM. Translational regulation of the mitochondrial genome following redistribution of mitochondrial microRNA in the diabetic heart. Circ Cardiovasc Genet 2015;8:785-802. ArticlePubMedPMC
- 137. Chen Q, Thompson J, Hu Y, Das A, Lesnefsky EJ. Metformin attenuates ER stress-induced mitochondrial dysfunction. Transl Res 2017;190:40-50. ArticlePubMedPMC
- 138. Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, Mattheus M, Devins T, Johansen OE, Woerle HJ, Broedl UC, Inzucchi SE. EMPA-REG OUTCOME Investigators. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med 2015;373:2117-2128. ArticlePubMed
- 139. Neal B, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, Erondu N, Shaw W, Law G, Desai M, Matthews DR. CANVAS Program Collaborative Group. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med 2017;377:644-657. ArticlePubMed
- 140. Wiviott SD, Raz I, Bonaca MP, Mosenzon O, Kato ET, Cahn A, Silverman MG, Zelniker TA, Kuder JF, Murphy SA, Bhatt DL, Leiter LA, McGuire DK, Wilding JPH, Ruff CT, Gause-Nilsson IAM, Fredriksson M, Johansson PA, Langkilde AM, Sabatine MS. DECLARE-TIMI 58 Investigators. Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N Engl J Med 2019;380:347-357. ArticlePubMed
- 141. Cosentino F, Grant PJ, Aboyans V, Bailey CJ, Ceriello A, Delgado V, Federici M, Filippatos G, Grobbee DE, Hansen TB, Huikuri HV, Johansson I, Juni P, Lettino M, Marx N, Mellbin LG, Ostgren CJ, Rocca B, Roffi M, Sattar N, Seferovic PM, Sousa-Uva M, Valensi P, Wheeler DC. ESC Scientific Document Group. 2019 ESC guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD. Eur Heart J 2020;41:255-323. PubMed
- 142. McMurray JJV, Solomon SD, Inzucchi SE, Kober L, Kosiborod MN, Martinez FA, Ponikowski P, Sabatine MS, Anand IS, Belohlavek J, Bohm M, Chiang CE, Chopra VK, de Boer RA, Desai AS, Diez M, Drozdz J, Dukat A, Ge J, Howlett JG, Katova T, Kitakaze M, Ljungman CEA, Merkely B, Nicolau JC, O'Meara E, Petrie MC, Vinh PN, Schou M, Tereshchenko S, Verma S, Held C, DeMets DL, Docherty KF, Jhund PS, Bengtsson O, Sjostrand M, Langkilde AM. DAPA-HF Trial Committees and Investigators. Dapagliflozin in patients with heart failure and reduced ejection fraction. N Engl J Med 2019;381:1995-2008. ArticlePubMed
- 143. Baartscheer A, Schumacher CA, Wust RC, Fiolet JW, Stienen GJ, Coronel R, Zuurbier CJ. Empagliflozin decreases myocardial cytoplasmic Na(+) through inhibition of the cardiac Na(+)/H(+) exchanger in rats and rabbits. Diabetologia 2017;60:568-573. ArticlePubMedPDF
- 144. Uthman L, Baartscheer A, Bleijlevens B, Schumacher CA, Fiolet JWT, Koeman A, Jancev M, Hollmann MW, Weber NC, Coronel R, Zuurbier CJ. Class effects of SGLT2 inhibitors in mouse cardiomyocytes and hearts: inhibition of Na(+)/H(+) exchanger, lowering of cytosolic Na(+) and vasodilation. Diabetologia 2018;61:722-726. ArticlePubMedPDF
- 145. Santos-Gallego CG, Garcia-Ropero A, Badimon J. Reply: benefits of empagliflozin beyond enhancing myocardial energetics? J Am Coll Cardiol 2019;74:826. PubMed
- 146. Baker HE, Kiel AM, Luebbe ST, Simon BR, Earl CC, Regmi A, Roell WC, Mather KJ, Tune JD, Goodwill AG. Inhibition of sodium-glucose cotransporter-2 preserves cardiac function during regional myocardial ischemia independent of alterations in myocardial substrate utilization. Basic Res Cardiol 2019;114:25. ArticlePubMedPMCPDF
- 147. Marso SP, Bain SC, Consoli A, Eliaschewitz FG, Jodar E, Leiter LA, Lingvay I, Rosenstock J, Seufert J, Warren ML, Woo V, Hansen O, Holst AG, Pettersson J, Vilsboll T. SUSTAIN-6 Investigators. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med 2016;375:1834-1844. ArticlePubMed
- 148. Marso SP, Daniels GH, Brown-Frandsen K, Kristensen P, Mann JF, Nauck MA, Nissen SE, Pocock S, Poulter NR, Ravn LS, Steinberg WM, Stockner M, Zinman B, Bergenstal RM, Buse JB. LEADER Steering Committee. LEADER Trial Investigators. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med 2016;375:311-322. ArticlePubMedPMC
- 149. Qiao H, Ren H, Du H, Zhang M, Xiong X, Lv R. Liraglutide repairs the infarcted heart: the role of the SIRT1/Parkin/mitophagy pathway. Mol Med Rep 2018;17:3722-3734. ArticlePubMedPMC
- 150. Ilkun O, Wilde N, Tuinei J, Pires KM, Zhu Y, Bugger H, Soto J, Wayment B, Olsen C, Litwin SE, Abel ED. Antioxidant treatment normalizes mitochondrial energetics and myocardial insulin sensitivity independently of changes in systemic metabolic homeostasis in a mouse model of the metabolic syndrome. J Mol Cell Cardiol 2015;85:104-116. ArticlePubMedPMC
- 151. Escribano-Lopez I, Diaz-Morales N, Rovira-Llopis S, de Maranon AM, Orden S, Alvarez A, Banuls C, Rocha M, Murphy MP, Hernandez-Mijares A, Victor VM. The mitochondria-targeted antioxidant MitoQ modulates oxidative stress, inflammation and leukocyte-endothelium interactions in leukocytes isolated from type 2 diabetic patients. Redox Biol 2016;10:200-205. ArticlePubMedPMC
- 152. Dietl A, Maack C. Targeting mitochondrial calcium handling and reactive oxygen species in heart failure. Curr Heart Fail Rep 2017;14:338-349. ArticlePubMedPDF
- 153. Baumgardt SL, Paterson M, Leucker TM, Fang J, Zhang DX, Bosnjak ZJ, Warltier DC, Kersten JR, Ge ZD. Chronic co-administration of sepiapterin and l-citrulline ameliorates diabetic cardiomyopathy and myocardial ischemia/reperfusion injury in obese type 2 diabetic mice. Circ Heart Fail 2016;9:e002424. PubMedPMC
- 154. Zhao P, Zhang J, Yin XG, Maharaj P, Narraindoo S, Cui LQ, Tang YS. The effect of trimetazidine on cardiac function in diabetic patients with idiopathic dilated cardiomyopathy. Life Sci 2013;92:633-638. ArticlePubMed
- 155. Rosano GM, Vitale C, Sposato B, Mercuro G, Fini M. Trimetazidine improves left ventricular function in diabetic patients with coronary artery disease: a double-blind placebo-controlled study. Cardiovasc Diabetol 2003;2:16. ArticlePubMedPMC
- 156. Zacharowski K, Blackburn B, Thiemermann C. Ranolazine, a partial fatty acid oxidation inhibitor, reduces myocardial infarct size and cardiac troponin T release in the rat. Eur J Pharmacol 2001;418:105-110. ArticlePubMed
- 157. Maier LS, Layug B, Karwatowska-Prokopczuk E, Belardinelli L, Lee S, Sander J, Lang C, Wachter R, Edelmann F, Hasenfuss G, Jacobshagen C. RAnoLazIne for the treatment of diastolic heart failure in patients with preserved ejection fraction: the RALI-DHF proof-of-concept study. JACC Heart Fail 2013;1:115-122. PubMed
- 158. Morrow DA, Scirica BM, Chaitman BR, McGuire DK, Murphy SA, Karwatowska-Prokopczuk E, McCabe CH, Braunwald E. MERLIN-TIMI 36 Investigators. Evaluation of the glycometabolic effects of ranolazine in patients with and without diabetes mellitus in the MERLIN-TIMI 36 randomized controlled trial. Circulation 2009;119:2032-2039. ArticlePubMed
- 159. Levelt E, Gulsin G, Neubauer S, McCann GP. Mechanisms in endocrinology: diabetic cardiomyopathy: pathophysiology and potential metabolic interventions state of the art review. Eur J Endocrinol 2018;178:R127-R139. ArticlePubMedPMC
- 160. Lee L, Campbell R, Scheuermann-Freestone M, Taylor R, Gunaruwan P, Williams L, Ashrafian H, Horowitz J, Fraser AG, Clarke K, Frenneaux M. Metabolic modulation with perhexiline in chronic heart failure: a randomized, controlled trial of short-term use of a novel treatment. Circulation 2005;112:3280-3288. ArticlePubMed
- 161. Beadle RM, Williams LK, Kuehl M, Bowater S, Abozguia K, Leyva F, Yousef Z, Wagenmakers AJ, Thies F, Horowitz J, Frenneaux MP. Improvement in cardiac energetics by perhexiline in heart failure due to dilated cardiomyopathy. JACC Heart Fail 2015;3:202-211. ArticlePubMed
- 162. Cappola TP. Perhexiline: lessons for heart failure therapeutics. JACC Heart Fail 2015;3:212-213. ArticlePubMed
- 163. Berthiaume JM, Kurdys JG, Muntean DM, Rosca MG. Mitochondrial NAD(+)/NADH redox state and diabetic cardiomyopathy. Antioxid Redox Signal 2019;30:375-398. ArticlePubMed
- 164. Martens CR, Denman BA, Mazzo MR, Armstrong ML, Reisdorph N, McQueen MB, Chonchol M, Seals DR. Chronic nicotinamide riboside supplementation is well-tolerated and elevates NAD(+) in healthy middle-aged and older adults. Nat Commun 2018;9:1286ArticlePubMedPMCPDF
- 165. Lee CF, Chavez JD, Garcia-Menendez L, Choi Y, Roe ND, Chiao YA, Edgar JS, Goo YA, Goodlett DR, Bruce JE, Tian R. Normalization of NAD+ redox balance as a therapy for heart failure. Circulation 2016;134:883-894. ArticlePubMedPMC
- 166. Yamamoto T, Byun J, Zhai P, Ikeda Y, Oka S, Sadoshima J. Nicotinamide mononucleotide, an intermediate of NAD+ synthesis, protects the heart from ischemia and reperfusion. PLoS One 2014;9:e98972. ArticlePubMedPMC
- 167. Waldman M, Nudelman V, Shainberg A, Abraham NG, Kornwoski R, Aravot D, Arad M, Hochhauser E. PARP-1 inhibition protects the diabetic heart through activation of SIRT1-PGC-1α axis. Exp Cell Res 2018;373:112-118. ArticlePubMed
REFERENCES
Fig. 1
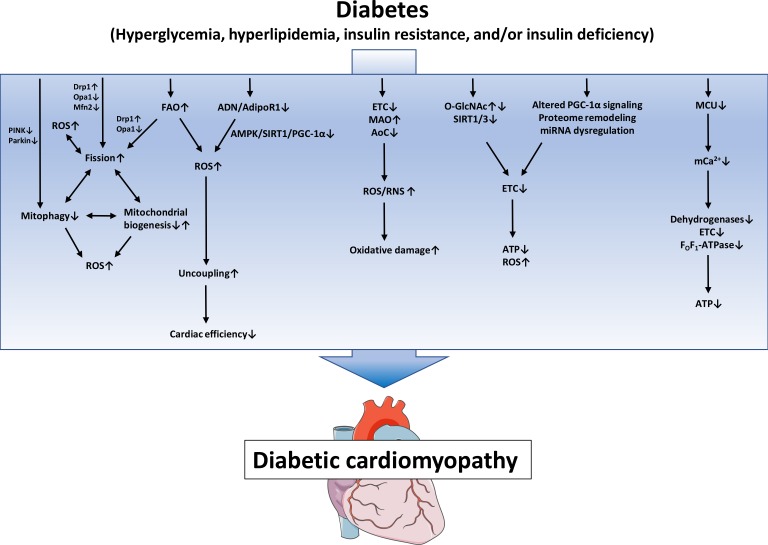
Proposed mechanisms of mitochondrial dysfunction in diabetic cardiomyopathy. Defects in the electron transport chain (ETC), increased monoamine oxidases (MAO) activity and decreased antioxidative capacity lead to increased reactive oxygen species/reactive nitrogen species (ROS/RNS) generation and subsequent oxidative damage. Posttranslational mechanisms like altered protein O-linked beta-N-acetylglucosamine glycosylation (O-GlcNAcylation) and increased protein acylation due to impaired SIRT activity, as well as mitochondrial proteome remodeling, impaired peroxisome proliferator-activated receptor gamma coactivator 1α (PGC-1α) signaling and miRNA dysregulation contribute to impaired ETC activity, ultimately leading to energy depletion and oxidative stress. Increased fatty acid oxidation (FAO) and/or impaired adiponectin (ADN)/adiponectin receptor 1 (AdipoR1) signaling may contribute to mitochondrial uncoupling and decreased cardiac efficiency. Increased mitochondrial fission, decreased mitophagy and altered mitochondrial biogenesis contribute to mitochondrial ROS and energy depletion and are interrelated mechanisms that may modulate each other. Impaired mitochondrial calcium uniporter (MCU) activity decreases mitochondrial Ca2+ uptake and thereby impairs activity of Ca2+-dependent dehydrogenases and oxidative phosphorylation. PINK1, phosphatase and tensin homolog-induced putative kinase 1; Drp1, dynamin-related protein 1; Opa1, optic atrophy 1; Mfn2, mitofusion 2; AMPK, adenosine monophosphate-activated protein kinase; SIRT1, sirtuin 1; AoC, antioxidative capacity; ATP, adenosine triphosphate.
Figure & Data
References
Citations
Citations to this article as recorded by 
- SIRT1: a promising therapeutic target in type 2 diabetes mellitus
Ainaz Mihanfar, Maryam Akbarzadeh, Saber Ghazizadeh Darband, Shirin Sadighparvar, Maryam Majidinia
Archives of Physiology and Biochemistry.2024; 130(1): 13. CrossRef - Effect of exercise on improving myocardial mitochondrial function in decreasing diabetic cardiomyopathy
Feng Zhang, Jian jian Lin, Hao nan Tian, Jun Wang
Experimental Physiology.2024; 109(2): 190. CrossRef - Cardioprotective effects of asiaticoside against diabetic cardiomyopathy: Activation of the AMPK/Nrf2 pathway
Chennian Xu, Lin Xia, Dengyue Xu, Yang Liu, Ping Jin, Mengen Zhai, Yu Mao, Yiwei Wang, Anguo Wen, Jian Yang, Lifang Yang
Journal of Cellular and Molecular Medicine.2024;[Epub] CrossRef - Ferroptosis: Mechanisms and role in diabetes mellitus and its complications
Pan Liu, Zhengdong Zhang, Yichen Cai, Zhaoying Li, Qian Zhou, Qiu Chen
Ageing Research Reviews.2024; 94: 102201. CrossRef - Salidroside Alleviates Myocardial Ischemia Reperfusion by Balancing Mitochondrial Homeostasis via Nrf2
Tingxu Yan, Xu Li, Xin Wang, Bosai He, Ying Jia, Wei Xiao, Rong He
Journal of Food Biochemistry.2024; 2024: 1. CrossRef - AGO2 Protects Against Diabetic Cardiomyopathy by Activating Mitochondrial Gene Translation
Jiabing Zhan, Kunying Jin, Rong Xie, Jiahui Fan, Yuyan Tang, Chen Chen, Huaping Li, Dao Wen Wang
Circulation.2024; 149(14): 1102. CrossRef - Cellular interplay between cardiomyocytes and non-myocytes in diabetic cardiomyopathy
Ren Jie Phang, Rebecca H Ritchie, Derek J Hausenloy, Jarmon G Lees, Shiang Y Lim
Cardiovascular Research.2023; 119(3): 668. CrossRef - Mitochondrial miRNA as epigenomic signatures: Visualizing aging-associated heart diseases through a new lens
Jasvinder Singh Bhatti, Naina Khullar, Rajesh Vijayvergiya, Umashanker Navik, Gurjit Kaur Bhatti, P. Hemachandra Reddy
Ageing Research Reviews.2023; 86: 101882. CrossRef - Eucommiae Folium and Active Compounds Protect Against Mitochondrial Dysfunction-Calcium Overload in Epileptic Hippocampal Neurons Through the Hypertrophic Cardiomyopathy Pathway
Shuai-nan Zhang, Hong-mei Li, Qi Liu, Xu-zhao Li, Wu-de Yang, Ying Zhou
Neurochemical Research.2023; 48(9): 2674. CrossRef - Speckle-tracking echocardiography provides sensitive measurements of subtle early alterations associated with cardiac dysfunction in T2DM rats
Yanchao Qi, Zhiyan Chen, Bingyan Guo, Zhe Liu, Lijie Wang, Suyun Liu, Lixiang Xue, Meifang Ma, Yajuan Yin, Yongjun Li, Gang Liu
BMC Cardiovascular Disorders.2023;[Epub] CrossRef - Novel insights into the role of mitochondria in diabetic cardiomyopathy: molecular mechanisms and potential treatments
Fumin Zhi, Qian Zhang, Li Liu, Xing Chang, Hongtao Xu
Cell Stress and Chaperones.2023; 28(6): 641. CrossRef - Transcription factor EB: A potential integrated network regulator in metabolic-associated cardiac injury
Weixing Wen, Haoxiao Zheng, Weiwen Li, Guolin Huang, Peng Chen, Xiaolin Zhu, Yue Cao, Jiahuan Li, Xiaohui Huang, Yuli Huang
Metabolism.2023; 147: 155662. CrossRef - Role of STIM1 in the Regulation of Cardiac Energy Substrate Preference
Panpan Liu, Zhuli Yang, Youjun Wang, Aomin Sun
International Journal of Molecular Sciences.2023; 24(17): 13188. CrossRef - Mitochondrial dysfunction at the crossroad of cardiovascular diseases and cancer
Carmine Rocca, Teresa Soda, Ernestina Marianna De Francesco, Marco Fiorillo, Francesco Moccia, Giuseppe Viglietto, Tommaso Angelone, Nicola Amodio
Journal of Translational Medicine.2023;[Epub] CrossRef - Triarylphosphonium compounds as effective vectors for mitochondria-targeted delivery systems: decoration strategies and prospects for clinical application
T. N. Pashirova, A. V. Nemtarev, E. B. Souto, V. F. Mironov
Russian Chemical Reviews.2023; 92(10): RCR5095. CrossRef - Moderate- and High-Intensity Endurance Training Alleviate Diabetes-Induced Cardiac Dysfunction in Rats
Sarah D’Haese, Maxim Verboven, Lize Evens, Dorien Deluyker, Ivo Lambrichts, BO Eijnde, Dominique Hansen, Virginie Bito
Nutrients.2023; 15(18): 3950. CrossRef - Alpha-lipoic acid enhances ischemic postconditioning-mediated improvement of myocardial infarction and apoptosis in diabetic rats with ischemia/reperfusion injury
Sanaz Gholami, Reza Badalzadeh, Alireza Alihemmati
Canadian Journal of Physiology and Pharmacology.2023; 101(12): 682. CrossRef - Concurrent diabetes and heart failure: interplay and novel therapeutic approaches
Qutuba G Karwi, Kim L Ho, Simran Pherwani, Ezra B Ketema, Qiuyu Sun, Gary D Lopaschuk
Cardiovascular Research.2022; 118(3): 686. CrossRef - Ca2+ mishandling and mitochondrial dysfunction: a converging road to prediabetic and diabetic cardiomyopathy
Carolina Jaquenod De Giusti, Julieta Palomeque, Alicia Mattiazzi
Pflügers Archiv - European Journal of Physiology.2022; 474(1): 33. CrossRef - Double-edge sword roles of iron in driving energy production versus instigating ferroptosis
Shuping Zhang, Wei Xin, Gregory J. Anderson, Ruibin Li, Ling Gao, Shuguang Chen, Jiajun Zhao, Sijin Liu
Cell Death & Disease.2022;[Epub] CrossRef - Differential remodelling of mitochondrial subpopulations and mitochondrial dysfunction are a feature of early stage diabetes
Bodour S. Rajab, Sarah Kassab, Connor D. Stonall, Hussam Daghistani, Stephen Gibbons, Mamas Mamas, David Smith, Aleksandr Mironov, Zainab AlBalawi, Yin Hua Zhang, Florence Baudoin, Min Zi, Sukhpal Prehar, Elizabeth J. Cartwright, Ashraf Kitmitto
Scientific Reports.2022;[Epub] CrossRef - GlyNAC (Glycine and N-Acetylcysteine) Supplementation Improves Impaired Mitochondrial Fuel Oxidation and Lowers Insulin Resistance in Patients with Type 2 Diabetes: Results of a Pilot Study
Rajagopal V. Sekhar
Antioxidants.2022; 11(1): 154. CrossRef - Glucose-derived posttranslational modification in cardiovascular disease
Michael Lehrke, Julia Moellmann, Florian Kahles, Nikolaus Marx
Molecular Aspects of Medicine.2022; 86: 101084. CrossRef - Mitochondrial Implications in Cardiovascular Aging and Diseases: The Specific Role of Mitochondrial Dynamics and Shifts
Anastasia V. Poznyak, Tatiana V. Kirichenko, Evgeny E. Borisov, Nikolay K. Shakhpazyan, Andrey G. Kartuesov, Alexander N. Orekhov
International Journal of Molecular Sciences.2022; 23(6): 2951. CrossRef - Editorial: Management of Diabetes and its Complications: A Focus on Endothelial Dysfunction
Shanhu Qiu, Jianhua Ma, Tongzhi Wu
Frontiers in Endocrinology.2022;[Epub] CrossRef - Alternative autophagy: mechanisms and roles in different diseases
Hong Feng, Nian Wang, Nan Zhang, Hai-han Liao
Cell Communication and Signaling.2022;[Epub] CrossRef - Mitochondria-Mediated Cardiovascular Benefits of Sodium-Glucose Co-Transporter 2 Inhibitors
Siarhei A. Dabravolski, Alexander D. Zhuravlev, Andrey G. Kartuesov, Evgeny E. Borisov, Vasily N. Sukhorukov, Alexander N. Orekhov
International Journal of Molecular Sciences.2022; 23(10): 5371. CrossRef - Animal Models of Dysregulated Cardiac Metabolism
Heiko Bugger, Nikole J. Byrne, E. Dale Abel
Circulation Research.2022; 130(12): 1965. CrossRef - Mitochondrial Dynamics and Mitophagy in Cardiometabolic Disease
Jianguo Lin, Jinlong Duan, Qingqing Wang, Siyu Xu, Simin Zhou, Kuiwu Yao
Frontiers in Cardiovascular Medicine.2022;[Epub] CrossRef - Perspectives for Forkhead box transcription factors in diabetic cardiomyopathy: Their therapeutic potential and possible effects of salvianolic acids
Ronghui Han, Hemeng Huang, Weiyi Xia, Jingjin Liu, Hui Luo, Jing Tang, Zhengyuan Xia
Frontiers in Cardiovascular Medicine.2022;[Epub] CrossRef - Molecular and cellular mechanisms in diabetic heart failure: Potential therapeutic targets
Misganaw Asmamaw Mengstie, Endeshaw Chekol Abebe, Awgichew Behaile Teklemariam, Anemut Tilahun Mulu, Assefa Agegnehu Teshome, Edgeit Abebe Zewde, Zelalem Tilahun Muche, Muluken Teshome Azezew
Frontiers in Endocrinology.2022;[Epub] CrossRef - Effect of Chronic Treatment with Uridine on Cardiac Mitochondrial Dysfunction in the C57BL/6 Mouse Model of High-Fat Diet–Streptozotocin-Induced Diabetes
Natalia V. Belosludtseva, Vlada S. Starinets, Irina B. Mikheeva, Maxim N. Belosludtsev, Mikhail V. Dubinin, Galina D. Mironova, Konstantin N. Belosludtsev
International Journal of Molecular Sciences.2022; 23(18): 10633. CrossRef - FGF21–Sirtuin 3 Axis Confers the Protective Effects of Exercise Against Diabetic Cardiomyopathy by Governing Mitochondrial Integrity
Leigang Jin, Leiluo Geng, Lei Ying, Lingling Shu, Kevin Ye, Ranyao Yang, Yan Liu, Yao Wang, Yin Cai, Xue Jiang, Qin Wang, Xingqun Yan, Boya Liao, Jie Liu, Fuyu Duan, Gary Sweeney, Connie Wai Hong Woo, Yu Wang, Zhengyuan Xia, Qizhou Lian, Aimin Xu
Circulation.2022; 146(20): 1537. CrossRef - Novel Insights Into Molecular Mechanism of Mitochondria in Diabetic Cardiomyopathy
Jing Bai, Chuanbin Liu, Pingjun Zhu, Yang Li
Frontiers in Physiology.2021;[Epub] CrossRef - Mitochondrial Dysfunction Increases Arrhythmic Triggers and Substrates; Potential Anti-arrhythmic Pharmacological Targets
Khalil Saadeh, Ibrahim Talal Fazmin
Frontiers in Cardiovascular Medicine.2021;[Epub] CrossRef - Prostaglandin E receptor subtype 4 protects against diabetic cardiomyopathy by modulating cardiac fatty acid metabolism via FOXO1/CD36 signalling
Fan Ying, Hao Liu, Eva Hoi Ching Tang, Ishan Lakhani, Ningning Liu, Zhengyuan Xia, Shiming Liu
Biochemical and Biophysical Research Communications.2021; 548: 196. CrossRef - Study of long non-coding RNA and mitochondrial dysfunction in diabetic rats
Haytham K. Sultan, Wael M. El-Ayat, Azza H. AbouGhalia, Noha N. Lasheen, Amr S. Moustafa
Tissue and Cell.2021; 71: 101516. CrossRef - A Role of Glucose Overload in Diabetic Cardiomyopathy in Nonhuman Primates
Xiu Wang, Shi Jin, Weina Hu, Gaetano Santulli
Journal of Diabetes Research.2021; 2021: 1. CrossRef - Dysregulation of circulating miRNAs promotes the pathogenesis of diabetes-induced cardiomyopathy
Uzair Ahmed, Usman Ali Ashfaq, Muhammad Qasim, Imtiaz Ahmad, Hafiz Usman Ahmad, Muhammad Tariq, Muhammad Shareef Masoud, Saba Khaliq, Muhammad Younas Khan Barozai
PLOS ONE.2021; 16(4): e0250773. CrossRef - MicroRNAs Regulating Mitochondrial Function in Cardiac Diseases
Guang-Qiong Zhang, Sheng-Quan Wang, Yan Chen, Ling-Yun Fu, Yi-Ni Xu, Ling Li, Ling Tao, Xiang-Chun Shen
Frontiers in Pharmacology.2021;[Epub] CrossRef - Effects of omega-3 fatty acids and metformin combination on diabetic cardiomyopathy in rats through autophagic pathway
Salma M. Eraky, Nehal M. Ramadan
The Journal of Nutritional Biochemistry.2021; 97: 108798. CrossRef - Adropin Alleviates Myocardial Fibrosis in Diabetic Cardiomyopathy Rats: A Preliminary Study
Mao Liu, Jiao Ai, Zhuang Shuai, Kai Tang, Zongyu Li, Yin Huang
Frontiers in Cardiovascular Medicine.2021;[Epub] CrossRef - Fibrosis of the diabetic heart: Clinical significance, molecular mechanisms, and therapeutic opportunities
Izabela Tuleta, Nikolaos G. Frangogiannis
Advanced Drug Delivery Reviews.2021; 176: 113904. CrossRef - Effect of the MPT Pore Inhibitor Alisporivir on the Development of Mitochondrial Dysfunction in the Heart Tissue of Diabetic Mice
Natalia V. Belosludtseva, Vlada S. Starinets, Irina B. Mikheeva, Dmitriy A. Serov, Maxim E. Astashev, Maxim N. Belosludtsev, Mikhail V. Dubinin, Konstantin N. Belosludtsev
Biology.2021; 10(9): 839. CrossRef - Characterisation of the Myocardial Mitochondria Structural and Functional Phenotype in a Murine Model of Diabetic Cardiomyopathy
Alex M. Parker, Mitchel Tate, Darnel Prakoso, Minh Deo, Andrew M. Willis, David M. Nash, Daniel G. Donner, Simon Crawford, Helen Kiriazis, Cesare Granata, Melinda T. Coughlan, Miles J. De Blasio, Rebecca H. Ritchie
Frontiers in Physiology.2021;[Epub] CrossRef - Radiation‑induced dysfunction of energy metabolism in the heart results in the fibrosis of cardiac tissues
Peng Xu, Yali Yi, Yijing Luo, Zhicheng Liu, Yilin Xu, Jing Cai, Zhimin Zeng, Anwen Liu
Molecular Medicine Reports.2021;[Epub] CrossRef - Heart Failure and Diabetes: Perspective of a Dangerous Association
Liliana E. Favaloro, Roxana D. Ratto, Carla Musso
Current Hypertension Reviews.2021; 17(2): 85. CrossRef - CTRP9 Mediates Protective Effects in Cardiomyocytes via AMPK- and Adiponectin Receptor-Mediated Induction of Anti-Oxidant Response
Bernd Niemann, Ling Li, Dorothee Siegler, Benedikt H. Siegler, Fabienne Knapp, Jakob Hanna, Muhammad Aslam, Michael Kracht, Rainer Schulz, Susanne Rohrbach
Cells.2020; 9(5): 1229. CrossRef - The Hippo Pathway Orchestrates Mitochondrial Quality Control: A Novel Focus on Cardiovascular Diseases
Ying Tan, Cai Lei, Huifang Tang, Xiao Zhu, Guanghui Yi
DNA and Cell Biology.2020; 39(9): 1494. CrossRef - Adeno-Associated Viral Transfer of Glyoxalase-1 Blunts Carbonyl and Oxidative Stresses in Hearts of Type 1 Diabetic Rats
Fadhel A. Alomar, Abdullah Al-Rubaish, Fahad Al-Muhanna, Amein K. Al-Ali, JoEllyn McMillan, Jaipaul Singh, Keshore R. Bidasee
Antioxidants.2020; 9(7): 592. CrossRef - Loss of function of transcription factor EB remodels lipid metabolism and cell death pathways in the cardiomyocyte
Purvi C. Trivedi, Jordan J. Bartlett, Angella Mercer, Logan Slade, Marc Surette, Andrea Ballabio, Stephane Flibotte, Bahira Hussein, Brian Rodrigues, Petra C. Kienesberger, Thomas Pulinilkunnil
Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease.2020; 1866(10): 165832. CrossRef - Rescue of myocardial energetic dysfunction in diabetes through the correction of mitochondrial hyperacetylation by honokiol
Matthew Kerr, Jack J. Miller, Dharendra Thapa, Sophie Stiewe, Kerstin N. Timm, Claudia N. Montes Aparicio, Iain Scott, Damian J. Tyler, Lisa C. Heather
JCI Insight.2020;[Epub] CrossRef - Diabetes Mellitus, Mitochondrial Dysfunction and Ca2+-Dependent Permeability Transition Pore
Konstantin N. Belosludtsev, Natalia V. Belosludtseva, Mikhail V. Dubinin
International Journal of Molecular Sciences.2020; 21(18): 6559. CrossRef - Effects of Standardized Green Tea Extract and Its Main Component, EGCG, on Mitochondrial Function and Contractile Performance of Healthy Rat Cardiomyocytes
Rocchina Vilella, Gianluca Sgarbi, Valeria Naponelli, Monia Savi, Leonardo Bocchi, Francesca Liuzzi, Riccardo Righetti, Federico Quaini, Caterina Frati, Saverio Bettuzzi, Giancarlo Solaini, Donatella Stilli, Federica Rizzi, Alessandra Baracca
Nutrients.2020; 12(10): 2949. CrossRef - Diabetic cardiomyopathy: definition, diagnosis criteria, treatment directions and prevention of heart failure
N. A. Koziolova, P. G. Karavaev, A. S. Veklich
South Russian Journal of Therapeutic Practice.2020; 1(2): 93. CrossRef - Adiponectin protects HL-1 cardiomyocytes against rotenone-induced cytotoxicity through AMPK activation
Biao Li, Baojian Zhang, Na Liu, Keke Wu, Yingxu Ma, Wanyun Zuo, Zuodong Ning, Yaozhong Liu, Chao Sun, Yichao Xiao, Tao Tu, Qiming Liu
Toxicology Letters.2020; 335: 82. CrossRef - Nutrient Sensor mTOR and OGT: Orchestrators of Organelle Homeostasis in Pancreatic β-Cells
Nicholas Esch, Seokwon Jo, Mackenzie Moore, Emilyn U. Alejandro, Yingke Xu
Journal of Diabetes Research.2020; 2020: 1. CrossRef - Where Does Metformin Stand in Modern Day Management of Type 2 Diabetes?
Ehtasham Ahmad, Jack Sargeant, Francesco Zaccardi, Kamlesh Khunti, David Webb, Melanie Davies
Pharmaceuticals.2020; 13(12): 427. CrossRef
 PubReader
PubReader Cite
Cite