- Current
- Browse
- Collections
-
For contributors
- For Authors
- Instructions to authors
- Article processing charge
- e-submission
- For Reviewers
- Instructions for reviewers
- How to become a reviewer
- Best reviewers
- For Readers
- Readership
- Subscription
- Permission guidelines
- About
- Editorial policy
Articles
- Page Path
- HOME > Diabetes Metab J > Volume 45(6); 2021 > Article
-
ReviewMetabolic risk/Epidemiology Obesity, Diabetes, and Increased Cancer Progression
-
Dae-Seok Kim1
 , Philipp E. Scherer1,2
, Philipp E. Scherer1,2 -
Diabetes & Metabolism Journal 2021;45(6):799-812.
DOI: https://doi.org/10.4093/dmj.2021.0077
Published online: November 22, 2021
1Touchstone Diabetes Center, Department of Internal Medicine, Dallas, TX, USA
2Department of Cell Biology, University of Texas Southwestern Medical Center, Dallas, TX, USA
-
Corresponding author: Philipp E. Scherer Touchstone Diabetes Center, University of Texas Southwestern Medical Center, 5323 Harry Hines Blvd, Dallas, TX, USA E-mail: philipp.scherer@utsouthwestern.edu
Copyright © 2021 Korean Diabetes Association
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- Rates of obesity and diabetes have increased significantly over the past decades and the prevalence is expected to continue to rise further in the coming years. Many observations suggest that obesity and diabetes are associated with an increased risk of developing several types of cancers, including liver, pancreatic, endometrial, colorectal, and post-menopausal breast cancer. The path towards developing obesity and diabetes is affected by multiple factors, including adipokines, inflammatory cytokines, growth hormones, insulin resistance, and hyperlipidemia. The metabolic abnormalities associated with changes in the levels of these factors in obesity and diabetes have the potential to significantly contribute to the development and progression of cancer through the regulation of distinct signaling pathways. Here, we highlight the cellular and molecular pathways that constitute the links between obesity, diabetes, cancer risk and mortality. This includes a description of the existing evidence supporting the obesity-driven morphological and functional alternations of cancer cells and adipocytes through complex interactions within the tumor microenvironment.
- The prevalence of obesity and diabetes has risen significantly for several decades and is expected to further increase gradually over the coming years [1]. Epidemiologically, individuals with obesity and diabetes are susceptible to an increased risk as well as a greater mortality rates for several types of cancers, such as endometrial, liver, pancreatic, colorectal, and breast cancer [2,3]. Therefore, understanding the molecular mechanisms underlying the epidemiological and mechanistic association between obesity, diabetes, and cancer has garnered significant attention as a therapeutic area of interest. Growing epidemiological evidence is hinting at a causal link between obesity, diabetes, and cancer [4-6]. Interestingly, it is the metabolic abnormalities associated with obesity and diabetes that may explain the link between metabolic dysregulation and cancer. Obesity leads to metabolic abnormalities of adipose tissue, affects the release of various hormones, adipokines, inflammatory cytokines, growth factors, enzymes, and free fatty acids [7,8]. These multiple metabolic substrates have been implicated as risk factors for cancer incidence and mortality [9-11]. In addition, the adipocyte-cancer cell crosstalk leads to morphological and functional changes in adipose tissue, resulting in changes to endocrine and paracrine signaling [10,12]. In turn, enhanced metabolic substrates released by altered adipose tissue physiology play a role in proliferation, invasion and metastasis of tumor cells [13]. Metabolic disturbances in type 2 diabetes mellitus (T2DM), such as hyperinsulinemia and dyslipidemia, have been proposed as causal links between diabetes and cancer [14,15]. High levels of insulin in hyperinsulinemia activate insulin/IGF-signaling and the subsequent activation of phosphatidylinositol 3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) and mitogen-activated protein kinase (MAPK) signaling pathways promote cancer cell growth, survival, motility, and drug resistance [16-19]. In addition, hyperlipidemia leads to increased levels of cholesterol and non-esterified fatty acids (NEFAs), which are responsible for activation of oncogenic signaling pathways, membrane synthesis, and adenosine triphosphate (ATP) [14]. Here, we highlight the molecular mechanisms that point to a causal link between development of cancer and metabolic abnormalities that are associated with obesity and diabetes.
INTRODUCTION
- Obesity alters the metabolic profile of adipose tissue and leads to the increased secretion of numerous hormones, adipokines, inflammatory cytokines, growth factors, enzymes, and free fatty acids [7,8]. These adipose tissue-specific secreted factors contribute to the initiation and progression of several cancer types by driving metabolic reprograming of cells [20-22]. There is growing awareness of the essential role of adipocytesecreted factors from obese adipose for the development and progression of cancer.
- Altered adipokine secretion
- As a major endocrine organ, adipose tissue produces and secretes a variety of bioactive polypeptides, referred to as adipokines [23,24]. More than 600 adipocyte-enriched secretory factors have been discovered to date and adipokines play a critical role in maintaining glucose and energy homeostasis, as well as a range of metabolic pathways through communication with other organs [25]. The excess expansion of adipose tissue in obesity alters adipokine secretion and promotes chronic low-grade inflammation, thereby contributing to the development of metabolic disorders, including obesity and T2DM (Fig. 1) [26]. In addition, the dysregulation of adipose tissuespecific adipokines has an impact on the cellular physiology of various tumor cells and affects cancer cell growth, proliferation [27], migration, invasion [28], epithelial-mesenchymal transition (EMT) [29], angiogenesis [29], metastasis [29], and development of multidrug-resistance [9,21,30,31]. These altered adipokine profiles are associated with an altered metabolic state and directly provide substrates to help cancer cells meet their energy demands for the various biological processes mentioned above. Two of the most abundant and well-studied adipokines, adiponectin and leptin, have garnered significant attention in the field of cancer [32]. A number of studies have shown that adiponectin plays a protective role against obesityassociated cancer development and progression [33]. Adiponectin, a 30-kDa adipokine encoded by the ADIPOQ gene, is involved in regulating insulin sensitivity, glucose levels, as well as fatty acid breakdown [34,35]. Low serum adiponectin levels are associated with chronic inflammation and metabolic disorders, including T2DM, obesity, cardiovascular disease, and cancer development [35]. Several studies have shown that serum adiponectin levels are inversely associated with the risk of developing several types of cancer [36-38]. For example, low levels of adiponectin are associated with increased cancer risk, while the higher adiponectin levels are linked to a decreased cancer risk, and reduced cancer progression [39]. However, these effects may be very indirect with high adiponectin levels being a correlate to lower body weight, thereby decreasing the risk of tumor development. Other studies have shown that once an oncogenic mutation has occurred within a tissue, adiponectin can exert a positive effect on tumor growth [40]. Several mechanisms have been proposed to explain how adiponectin regulates cellular signaling pathways in cancer. Upon binding to its receptors, AdipoR1 and AdipoR2, and the possible engagement of the adaptor protein, phosphotyrosine interacting with PH domain and leucine zipper 1 (APPL1), adiponectin regulates multiple downstream signaling pathways, including adenosine monophosphate-activated protein kinase (AMPK), mTOR, PI3K/protein kinase B (AKT), MAPK, peroxisome proliferator-activated receptor γ (PPARγ), signal transducer and activator of transcription 3 (STAT3), and nuclear factor-κB (NF-κB) [41-45]. Further studies are required to resolve the heterogeneous effect of adiponectin on tumor growth. Leptin is a 16-kDa multifunctional peptide hormone produced mainly by adipocytes and regulates food intake and energy expenditure to maintain body weight [46]. Plasma leptin concentrations increase in proportion to body fat mass, and elevated circulating leptin levels are a hallmark of obesity [32]. Growing evidence suggests that excess leptin and overexpression of its receptor (Ob-R) impacts the signaling pathways involved in cell proliferation, migration, invasion, metastasis, and EMT in breast cancer [47]. Leptin binds to Ob-R on breast cancer cells and within tumor tissue enhances several tumor cell responses via aberrant activation of multiple signaling pathways, such as the activation of the MAPK kinase (MEK)/extracellular signal-regulated kinase (ERK)1/2, Jak/STAT3, and PI3K/Akt signaling pathways [47]. Leptin and Ob-R are known to be expressed in estrogen receptor (ER)-positive breast cancer types and enhance ERα-dependent transcription to promote cell proliferation [48]. Interestingly, recent studies have begun to reveal that Ob-R is also highly expressed in hormone receptor-negative breast cancers, indicating that leptin and Ob-R may have a distinct role in the context of hormonereceptor negative breast cancers [49]. The functional interactions between leptin and Ob-R in the regulation of hormone receptor-negative breast cancers are highly intriguing and promise to be an excellent area of intervention in future studies. In addition, the generation and secretion of leptin also increased in epithelial tumor cells and tumor-associated stromal cells, suggesting that the production of leptin is not limited to adipocytes in the tumor microenvironment [50]. Importantly, multiple clinical studies have highlighted that elevated serum leptin levels are associated with cancer progression, metastasis, and poorer prognosis in patients with various other cancer types [47]. Therefore, given their key roles in various cancers, leptin and Ob-R are attractive therapeutic targets. As such, the inhibition of the leptin/Ob-R interaction and their clinical applications have attracted substantial interest and opened another avenue for treatment advances with the potential to dramatically impact breast cancer.
- Insulin resistance
- Insulin is a hormone that is produced by β-cells in the pancreas and is primarily responsible for stimulating glucose uptake and storage to regulate glucose levels in blood [51]. Insulin also is involved in evolutionarily conserved pathways, such as cell growth, proliferation, and differentiation as well as protein and lipid synthesis, RNA and DNA synthesis. Mechanically, insulin binds to the insulin receptor (IR), a tyrosine kinase, that stimulates glucose uptake into metabolically active tissues such as skeletal muscle, adipose tissue and liver through enhanced translocation of the insulin-dependent glucose carrier 4 (glucose transporter 4 [GLUT4]) to the plasma membrane. Insulin resistance is clinically defined as the state in which a given concentration of insulin exerts a biological effect lower than expected. Impaired insulin-stimulated glucose uptake as well as reduced glucose oxidation and glycogen synthesis are known to be associated with insulin resistance [51]. Frequently, obesity is associated with development of insulin resistance (Fig. 1). Obesity may very well cause insulin resistance through promoting chronic inflammation in adipose tissue, and by increasing insulin secretion in the system, thereby activating multiple growth pathways [7]. However, insulin per se can also cause obesity due to its nature as a potently anabolic hormone. Chronic inflammation, excess insulin secretion and hyperactivation of growth pathways are closely associated with tumor development and progression [52]. Thus, obesity-induced insulin resistance plays a pathogenic role in contributing increased higher cancer incidence and cancer‐specific mortality.
- Increased inflammatory cytokines
- Obesity-associated chronic low-grade inflammation is known to be major factor in the development of metabolic disease and several types of cancer [9]. Chronic low-grade inflammation is characterized by increased infiltration and activation of immune cells along with increased local and systemic cytokine levels, leading to subsequent immune dysregulation in adipose tissue, liver, pancreas, skeletal muscle, and brain. Activation of several metabolic signaling pathways including IKKβ/NF-κB and c-Jun N-terminal kinase (JNK) pathways by excess nutrients leads to initiation of obesity-associated inflammation and a subsequent chronic low-grade inflammatory response (Fig. 1) [9]. In healthy and lean adipose tissue, M2 macrophages secrete antiinflammatory interleukin 10 (IL-10) cytokines, and M2 marker genes such as arginase 1 (Arg-1), fizzled, and Ym1 to maintain adipose tissue homeostasis [7]. In contrast to lean white adipose tissue, macrophages are recruited and infiltrated to white adipose tissue under obesogenic conditions and contribute to insulin resistance through promoting the secretion of proinflammatory cytokines, such as tumor necrosis factor-α (TNF-α), IL-6, IL-1β, interferon γ (IFN-γ). Obese white adipose tissue can also shift the polarization of macrophages. During the course of obesity, the levels of the broadly defined pro-inflammatory M1 macrophages increase, leading to adipose tissue inflammation and insulin resistance by inhibiting IR signaling [7,53]. Enhanced secretion of macrophage-derived pro-inflammatory cytokines in obese adipose tissue is a major contributor to the pathogenesis of tumor development by activating several signaling pathways, such as the JAK/STAT pathway [9-11,54].
- Hormonal disorders
- Aromatase, belongs to cytochrome P450 family of monooxygenases, is an enzyme responsible for metabolism and cholesterol/steroid synthesis including estrogen biosynthesis [55]. Obesity has been associated with abnormally high aromatase expression in mammary tissues, which is synthesized in undifferentiated pre-adipocytes and adipose fibroblasts in postmenopausal women (Fig. 1) [2]. Obesity is associated with elevated expression and secretion of proinflammatory cytokines, such as TNF-α and IL-6 in adipose tissue, which contributes to increased aromatase expression in adipose tissue [56]. Elevated expression of aromatase in breast adipose tissue promotes the conversion of androgen to estrogen, leading to increased local estradiol concentrations and ERs activation [57-60]. Estrogens exert their biological functions via ERs that are ligand-dependent transcription factors that activate genes that are involved in cell proliferation, differentiation, apoptosis, and cell migration [61]. Dysregulated actions of ERα signaling are associated with breast cancer initiation and development [61,62]. ER+ tumors typically receive their growth signal from estrogen to promote cellular growth. In postmenopausal women, estrogen is mainly produced in peripheral tissues via the enzyme aromatase, thus inhibition of aromatase activity is often an effective treatment for ER+ tumors. Collectively, systemic and local estrogen production by aromatase in obese individuals contributes to the risk of hormone-dependent cancers. Therefore, inhibition of aromatase activity in the breast tissue is an established and effective treatment to curb the growth of estrogenreceptor-positive breast cancers.
- Alternations in the tumor microenvironment
- The mutual interactions between tumor cells and their adjacent microenvironment that result in a loss of tissue homeostasis imposed by changes in tissue architecture and polarity [63,64]. The tumor microenvironment has been recognized as a key contributor to many aspects of cancer development [65]. The extracellular matrix (ECM) and hypoxia have been widely demonstrated to play a critical role in the regulation of the tumor microenvironment. The main components of the ECM include various proteoglycans and fibrous proteins such as collagens, fibronectin, elastin, hyaluronan, and laminin. The ECM is one of the essential components of the tumor microenvironment, providing structural and mechanical support to tumor cells [66-68]. It is well established that tumor-driven ECM remodeling alters the biochemical and mechanical properties of the tumor microenvironment to create a favorable microenvironment for tumor cell growth, migration and metastatic progression [69-71]. The high oxygen demand of rapidly proliferating cancer cells causes an insufficient oxygen supply to the tumor, which becomes hypoxic [72]. Hypoxia is a well-known characteristic of the tumor microenvironment and promotes tumor angiogenesis, metastasis, reprogramming of metabolism, and resistance to therapy [73]. Emerging studies suggest that hypoxia also inhibits anti-tumor immune cells, which allows tumor cells to evade the immune system, thus establishing an immunosuppressive tumor microenvironment [74-77].
- Obesity can influence the tumor microenvironment through dysfunctional adipose tissue and altered extracellular signals, thus promoting tumor growth, proliferation, angiogenesis, invasion, migration, and metastasis in breast cancer (Fig. 1) [13,78]. During obesity, vascular dysfunction is impaired and creates pockets of hypoxia. Adipose tissue hypoxia establishes a highly proinflammatory microenvironment, which is a favorable microenvironment for tumor promotion [65]. Dysfunctional adipose tissue leads to dysregulated adipokine production, preferentially releasing proinflammatory adipokines [23]. This adipokine imbalance is closely associated with insulin resistance, lipolysis and pro-inflammatory signaling pathways, contributing to a favorable microenvironment for tumor growth and progression. In addition, macrophage infiltration into obese adipose tissues promote ECM remodeling through elevation of several ECM components, thereby providing a tumor-friendly environment [21].
- Activation of mitogenic signaling pathways
- Obesity induces multiple intracellular signaling pathways through elevation of various signaling molecules including insulin, leptin and adiponectin (Fig. 1) [8]. Insulin and insulin-like growth factor-1 binding to their receptors (IR/IGF-1R), activating the PI3K/AKT/mTOR pathway, driving cell proliferation, invasion and metastasis [79-81]. Higher circulating levels of leptin secreted by obese adipose tissue binds to its cognate receptor (Ob-R), activating multiple signaling pathways, such as the JAK-STAT, PI3-kinase-AKT, and MAPK pathways, resulting in increased proliferation, migration and invasion of breast cancer cells [82,83]. In addition, bidirectional crosstalk between leptin and IGF-1 signaling plays an important role in obesity-associated breast cancer progression through epidermal growth factor receptor (EGFR) transactivation [84]. Adiponectin is known to inhibit tumor development and growth through activation of AMPK and subsequent downregulation of the MAPK pathway [36,85]. In addition, elevated glucose levels potentiate Wnt/β-catenin signaling through promoting the nuclear translocation of β-catenin [86]. Activation of the Wnt/β-catenin pathway enhances hypoxia-inducible factor 1α (HIF1α)-activated gene expression, thereby promoting cell survival and angiogenesis during hypoxia.
DYSFUNCTIONAL ADIPOSE TISSUE AND CANCER DEVELOPMENT
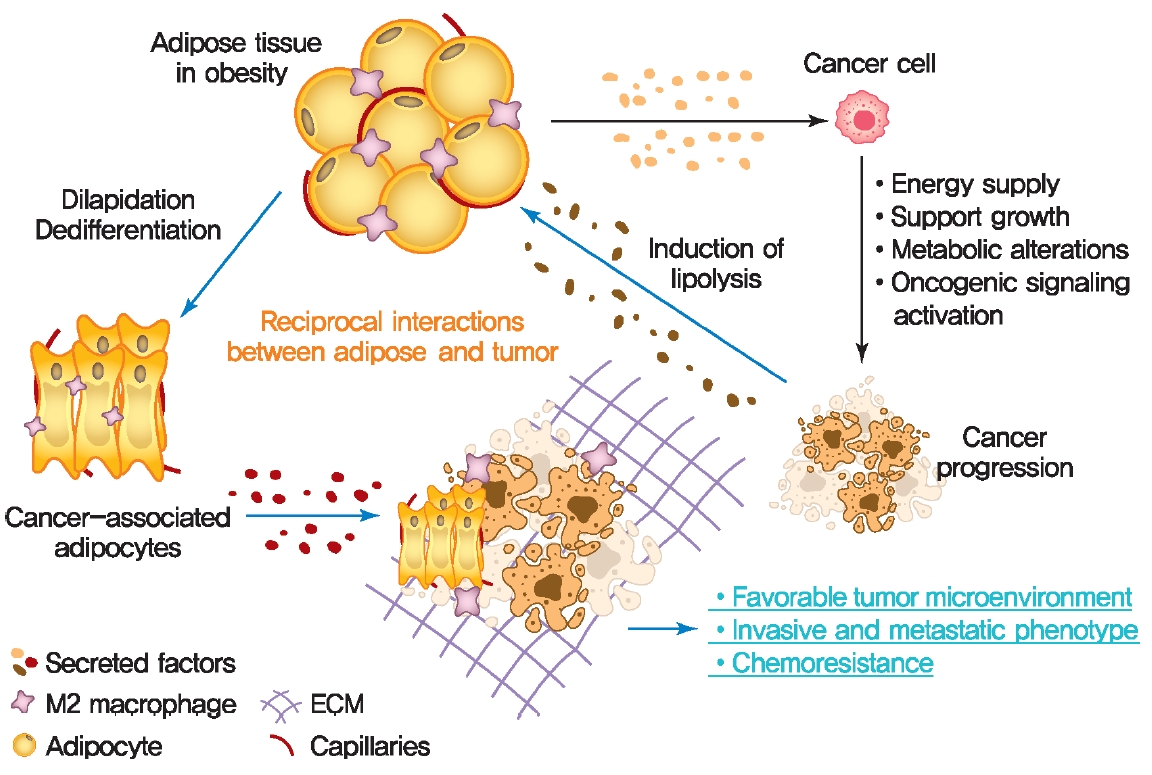
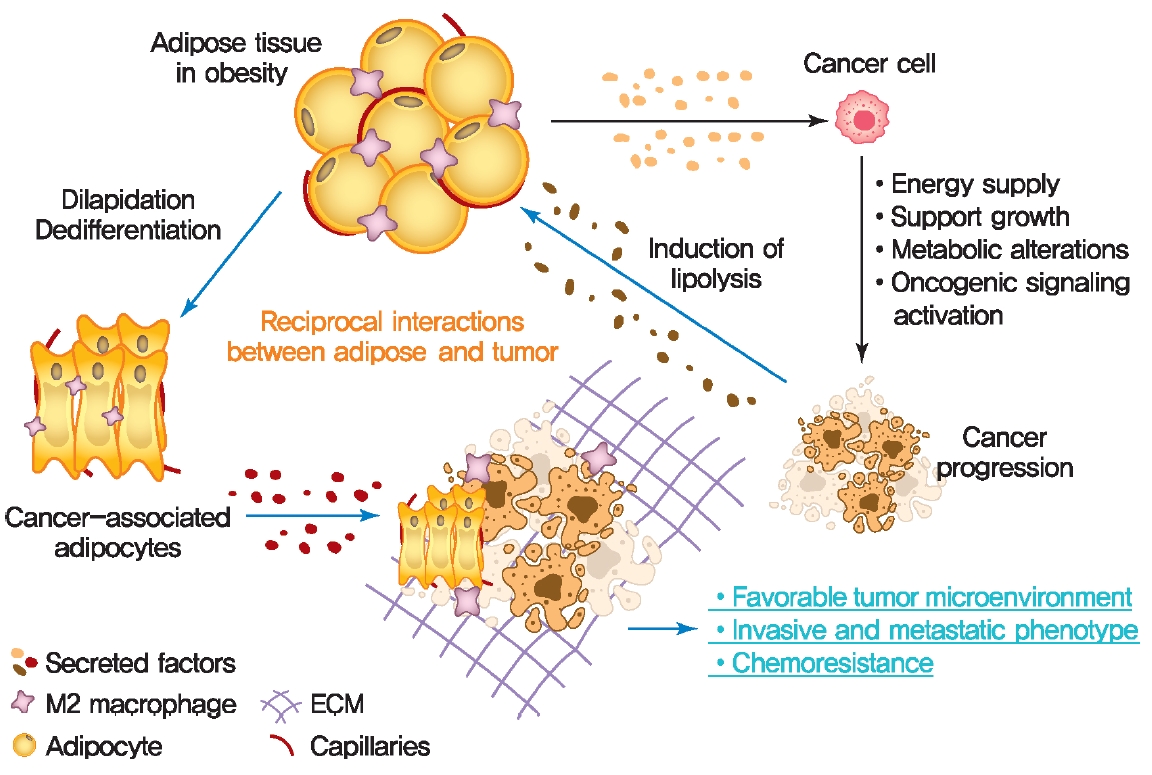
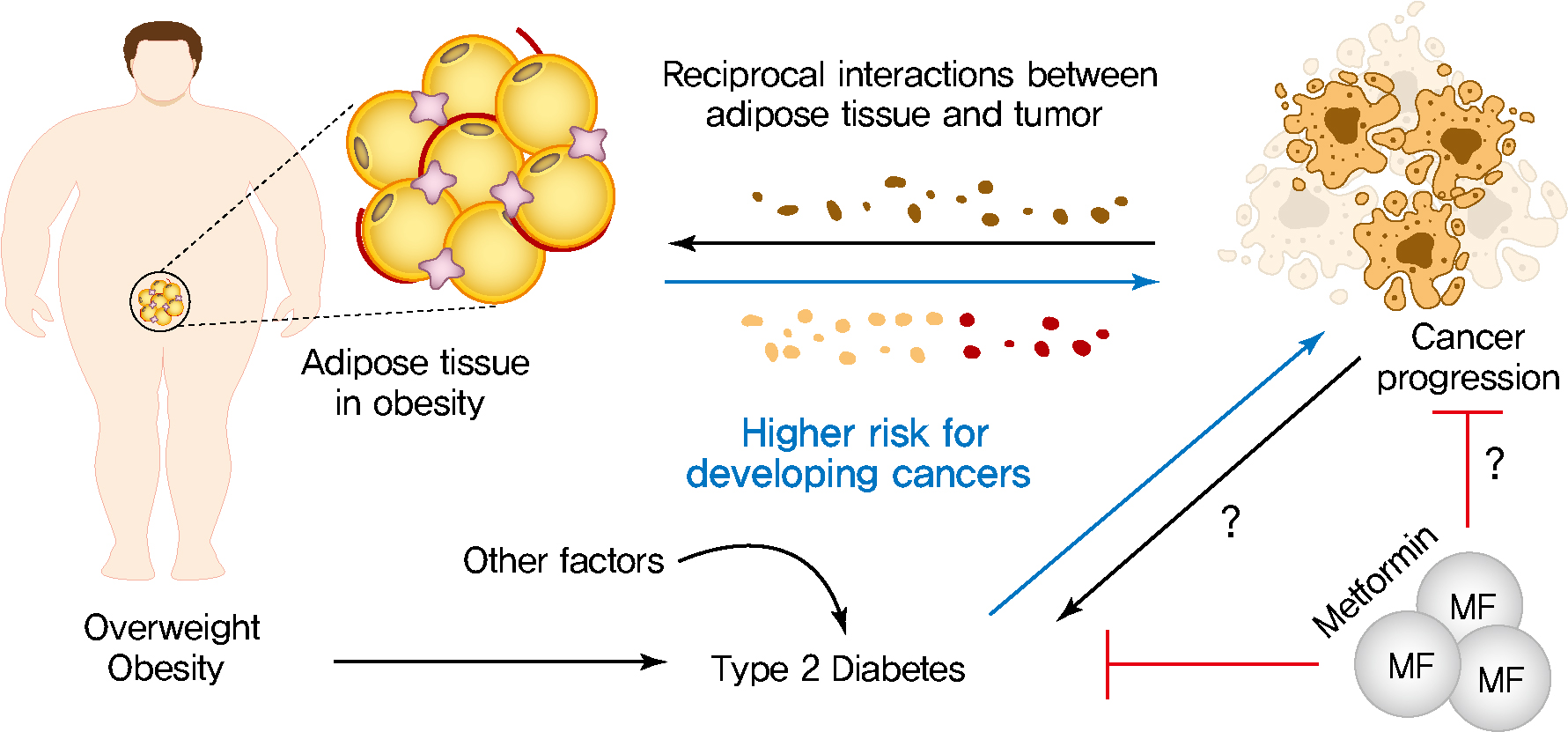
- The crosstalk between adipose tissue and tumors in the obese state
- Altered adipose tissue homeostasis in obesity can provide factors such as hormones, adipokines, and cytokines that assist cancer cells in acquiring the increased metabolic and energy demands mentioned above (Fig. 2) [7,9,56]. The crosstalk between cancer cells and adipocytes within the tumor microenvironment leads to further morphological and functional alteration of both cell types [10,78,87]. During interactions with cancer cells, adipocytes acquire phenotypic changes, including delipidation through multiple bioactive factors released by the cancer cells, resulting in a significantly decreased the expression of adipocyte-specific genes such as adiponectin, leptin, fatty acid-binding protein-4 (FABP4) [56]. Compared to normal adipocytes, this fibroblast-like phenotype of adipocytes is referred to as cancer-associated adipocytes (CAAs) [78]. Continuous stimulation of CAAs by paracrine signals from cancer cells leads to secretion of free fatty acids, inflammatory cytokines, adipokines, and growth factors, establishing a favorable tumor microenvironment which is an integral part of cancer development. Therefore, a greater understanding of functional crosstalk between CAA and cancer cell and new strategies for blocking this interaction may be an effective/attractive therapeutic target for treatment of cancer.
- Cancer metabolic reprogramming by adipokines
- Cancer cells undergo reprogramming of glucose metabolism by increasing glucose uptake and glycolytic activity in the presence of oxygen (referred to as the Warburg effect) [88]. Growing evidence suggest that this glucose metabolic reprograming is driven by various adipose tissue-specific secreted factors. For example, leptin is known to promote glycolysis by stimulating key glycolytic enzymes and GLUTs, hexokinase, the M2 isoform of pyruvate kinase (PKM2), lactate dehydrogenase A (LDHA), and glucose-6-phosphate dehydrogenase (G6PDH) through PI3K/Akt activation [89-91]. Recent studies have revealed that p62 deficiency in adipose tissue supports nutrient availability for cancer cells through inhibition of energy-utilizing pathways in adipocytes [92]. Loss of p62 from adipose tissue leads to increased levels of osteopontin, an adipokine that subsequently enhances tumor cell fatty acid oxidation. Fatty acid oxidation provides the energy necessary for cancer cells to support tumorigenesis and cancer progression. These observations indicate a central role of adipose tissue-specific secreted factors in the regulation of cancer cell metabolic reprogramming (Fig. 2).
INTERACTION BETWEEN OBESITY AND CANCER
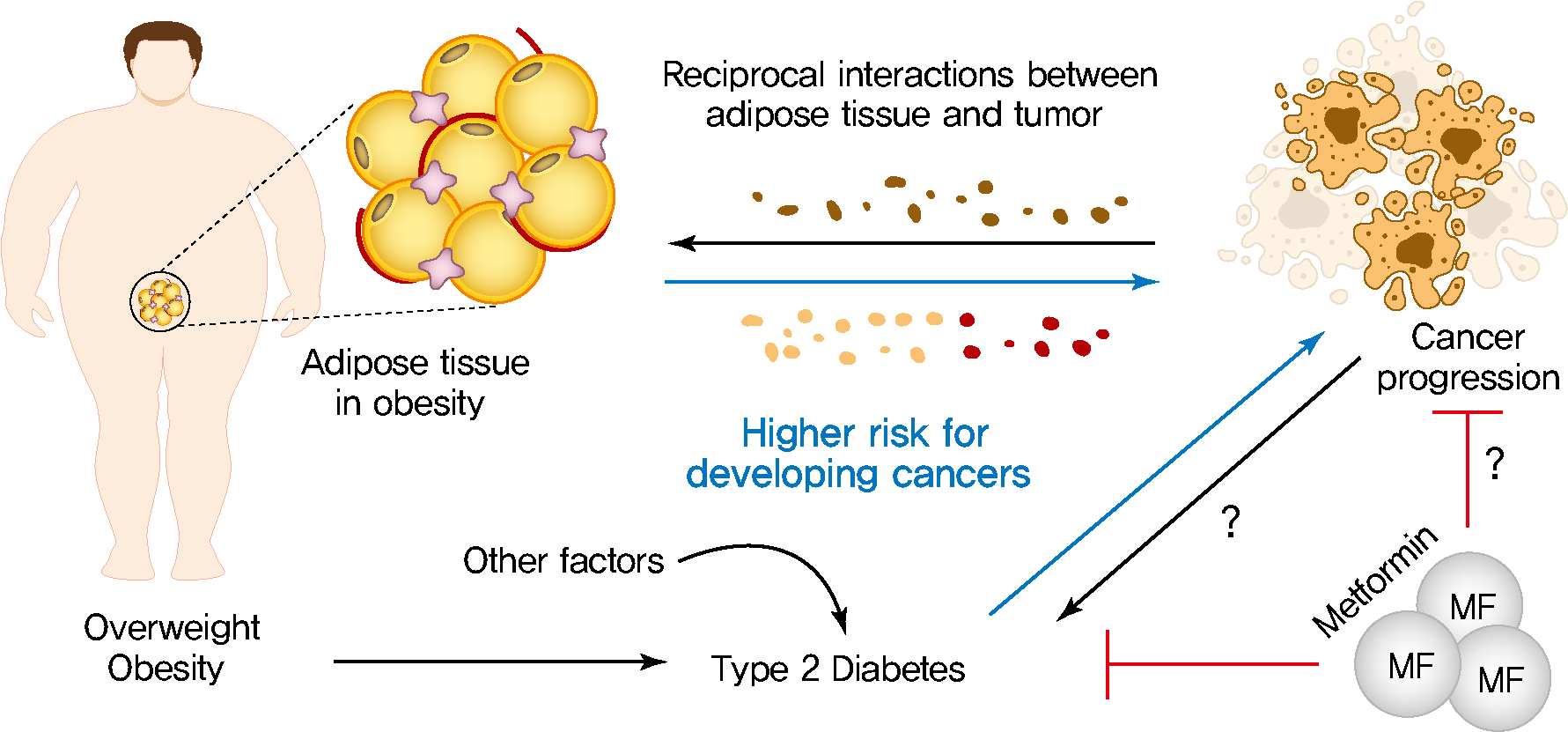
- Diabetes mellitus (DM) is one of the most prevalent chronic metabolic disorder characterized by hyperglycemia [3]. Dysfunction of pancreatic insulin-producing β-cells and insulin resistance mainly leads to hyperglycemia, resulting in increased risk of T2DM development [93]. T2DM is most common form of DM, which accounts for around 90% of all cases of diabetes [93]. T2DM may be associated with increased risk, accelerated progression and greater mortality rates of several types of cancer such as liver, pancreatic, endometrial, colorectal, and breast cancer [3,94,95]. Anti-diabetic drugs, such as metformin, have recently attracted considerable interest and opened a promising avenue of research that has the potential for the treatment of breast and colorectal cancers due to their proposed anti-cancer properties [95-98]. In the following section, we discuss the various mechanism by which metabolic disturbances in T2DM, such as dyslipidemia, hyperinsulinemia, and hyperglycemia, may lead to the development of breast cancer.
- Hyperinsulinemia
- Hyperinsulinemia is characterized by chronically elevated insulin in the blood due to dysregulated insulin secretion and/or clearance [93]. Higher levels of the IR and increased circulating insulin are important factors that drive the risk of developing several cancers in T2DM patients (Fig. 3) [99-102]. Binding of insulin to IR leads to activating tyrosine kinase activity of the IR, resulting in tyrosine phosphorylation of IR substrate and subsequent activation of PI3K/Akt pathway which is responsible for most metabolic and mitogenic effects of insulin [103]. Activation of IR/PI3K/Akt signaling pathway directly phosphorylates and thus activates mTOR. mTOR then activates its downstream signaling pathway, which is involved in regulating cancer cell survival, proliferation, invasion, migration, differentiation, angiogenesis, and metastasis [103,104]. In addition, activation of IR/PI3K/Akt signaling pathway stimulates β-catenin translocation into the nucleus, increases the levels of vascular endothelial growth factor (VEGF), thus impacts the various tumor cell behaviors [105,106]. The IGFs are peptide hormones with high structural and functional similarity to insulin [103]. Binding of IGFs to their receptors (IGFRs) activates the intrinsic tyrosine kinase activity of receptor and initiates an intracellular signaling cascade, thereby play important roles in growth and development of many organs. Insulin and IGFs are able to bind to each other’s receptors, IR and IGFRs, and thus activate common downstream signaling including PI3K/Akt pathway and MAPK pathway [103,107]. High levels of insulin can lead to binding and activation of IGFRs signaling as well as an increase in the levels of bioactive IGFs, thus playing an essential role in cancer cell growth, survival, motility, and drug resistance.
- Hyperlipidemia
- Dyslipidemia is often observed in patients with obesity, T2DM, and cardiovascular diseases [108]. Hyperlipidemia is characterized by alterations in circulating lipids with elevated low-density lipoprotein cholesterol, high levels of triglycerides, and low high-density lipoprotein cholesterol. Increased levels of cholesterol and NEFAs can indirectly act as signaling molecules involved in activation of intracellular signaling pathways such as the Akt/mTOR and Akt/glycogen synthase kinase 3β (GSK3β)/β-catenin oncogenic pathway (Fig. 3) [14]. 27-Hydroxycholesterol (27HC) is a primary metabolite of cholesterol, generated upon exposure to cytochrome P450 oxidase sterol 27-hydroxylase A1 (CYP27A1), an enzyme involved in regulating cellular cholesterol homeostasis [18]. 27HC acts as an ER agonist to activate ER related signaling mechanisms, such as the Akt/mTOR pathway or Akt/GSK3β/β-catenin, thereby stimulating cell proliferation and protein synthesis in ER-positive breast cancers (Fig. 3) [109,110]. High levels of CYP27A1 expression correlate with high-grade tumors in human breast cancer specimens, and inhibition of CYP27A1 reduces tumor growth in hormone-dependent breast cancers [111,112]. Interestingly, the expression of 27HC is different in various obesity models such as diet-induced obesity, db/db mice, and ob/ob mice. Therefore, future studies should address the how the dynamic changes of 27HC expression under different obesity conditions affect ER-positive breast cancer development [113]. The protein kinase C θ (PKCθ) is activated by elevated levels of serum NEFAs, which causes activation of Akt/mTOR and Akt/GSK3β/β-catenin oncogenic pathway [114,115]. Therefore, reducing the circulating levels of cholesterol and NEFAs and interfering activity of CYP27A1 and PKCθ present potential strategies for curbing breast cancer growth. Cholesterol is an integral component of membranes and lipid rafts in highly proliferative cancer cells [116]. In addition to the regulation of cellular signaling pathways, cholesterol also serves as the precursor for steroid hormone synthesis, which drives the initiation and progression of several cancers, indicating that cancer cells require cholesterol for tumor growth and survival [117]. Elevated NEFA levels in cancer are responsible for generation of ceramides. Ceramide glycosylation by glucosyl ceramide synthase (GCS) confers upon cancer cells multidrug resistance to cytotoxic anticancer agents [14,118]. In addition, NEFAs are also involved in ATP generation through the β oxidative pathway as well as synthesis of signaling and membrane lipids to support cancer cell proliferation and tumor growth [14].
- The potential effect of diabetes medication metformin in cancer prevention
- A series of studies have assessed whether the use of insulinsensitizing medications, such as metformin [95-98,119-124] and thiazolidinediones (TZDs) [119,120,125,126], may lower the incidence of cancer in diabetic patients (Fig. 3). Metformin is a first-line pharmacological treatment for T2DM and reduces hepatic glucose production by decreasing hepatic gluconeogenesis, resulting in increasing insulin sensitivity [127]. Multiple mechanistic studies with metformin in vitro and in animal models have elucidated the molecular mechanisms that underlie the action of metformin [127]. In particular, one major effect of metformin has been proposed to be the suppression of mTOR signaling pathway. Metformin reduces the circulating levels of insulin and IGF-1 in peripheral blood and activates liver kinase B1 (LKB1)/AMPK signaling pathways, leading to inhibition of the mTOR pathway, thus reducing cell proliferation, protein translation, and insulin levels [128]. However, the possible role of metformin as preventive agent against cancer in patients remains controversial and now is being extensively studied. A multiple population-based cohort study in patients with diabetes indicated that metformin therapy was not significantly associated with a reduced risk of cancer among patients with diabetes [129-131]. Further prospective mechanistic and large population-based cohort studies will be required to evaluate the effect of metformin therapy in cancer incidence.
- Thiazolidinediones
- TZDs, PPARγ agonists, are insulin-sensitizing medications that reduce insulin resistance and decrease hepatic glucose production agents in T2DM [132]. In addition to their essential metabolic actions in T2DM, TZDs have been shown to exert anti-tumor effects by affecting the cell cycle, apoptosis and cell differentiation in several types of cancer including breast and colon cancer [133]. However, multiple clinical studies revealed that treatment with TZDs was not associated with a significantly anti-neoplastic effect in several cancers [134-136]. Therefore, further large-scale prospective studies will likely need to resolve these inconsistencies.
T2DM AND CANCER DEVELOPMENT
- The growing prevalence of obesity and diabetes are closely linked to an increased incidence and mortality of many types of cancer [2-4,9,56,94]. The multiple metabolic factors and disorders linking obesity and T2DM with cancer that are broadly discussed in this review may be important for determining the therapeutic potential of cancer treatment. Altered adipose tissue metabolism in obesity results in altered levels of multiple factors, such as hormones, adipokines, inflammatory cytokines, growth factors, enzymes, and free fatty acids to help cancer cells satisfy their metabolic and energy demands [7,13,24]. Adipocytes also acquire phenotypic changes through multiple bioactive factors released by surrounding cancer cells, resulting in significantly increased secretion of many metabolic substrates [30,78,87]. This adipocyte/cancer cell crosstalk within the tumor microenvironment leads to further morphological and functional alterations of both cell types, which is gradually being recognized as an integral part of cancer development and progression. However, many important questions remain regarding the molecular fingerprint and the biological roles of such a crosstalk in promoting cancer development and progression. Therefore, (1) a more in-depth understanding of how adipocytes interact with tumor cells and contribute to cancer development and progression is required; (2) identification of specific targets that can serve as promising avenues to limit tumor proliferation need to be identified; (3) determination of new strategies to block this interaction could be an effective/attractive therapeutic strategy in the treatment of cancers. T2DM is the most common form of diabetes that appears to be associated with an increased risk of several types of cancer due to its associations with multiple metabolic disturbances [94,95]. Various metabolic disturbances involved in the development of diabetes, such as dyslipidemia and hyperinsulinemia, are well established to lead to the development of a cancer-conducive microenvironment. Based on these observations, a series of studies suggest that the use of anti-diabetic drugs, e.g., metformin, may have the potential to reduce the cancer incidence and/or mortality in diabetic patients. Mechanistically, metformin reduces the circulating levels of insulin and IGF-1, thus inhibiting the mTOR pathway and subsequent cancer cell proliferation [128]. Given the potential of metformin for cancer therapy, metformin has attracted increased attention and opened new avenues in cancer treatment due to their proposed anti-cancer properties. In contrast, a recent a population-based cohort study indicated that metformin use was not associated with significantly decreased risk of cancer among patients with diabetes [129-131]. Based on large population-based cohort studies, further insights into the potential utility of metformin will be required to evaluate the effect of metformin therapy in cancer incidence.
CONCLUSIONS
-
CONFLICTS OF INTEREST
No potential conflict of interest relevant to this article was reported.
-
FUNDING
Authors were supported by US National Institutes of Health grants R01-DK55758, R01-DK- 127274, R01-DK099110, RC2-DK118620 and P01-AG051459 to Philipp E. Scherer.
NOTES
-
Acknowledgements
- We thank Dr. Christine Kusminski for comments and extensive editing of the manuscript.


- 1. Pillon NJ, Loos RJ, Marshall SM, Zierath JR. Metabolic consequences of obesity and type 2 diabetes: balancing genes and environment for personalized care. Cell 2021;184:1530-44.ArticlePubMedPMC
- 2. Gallagher EJ, LeRoith D. Obesity and diabetes: the increased risk of cancer and cancer-related mortality. Physiol Rev 2015;95:727-48.ArticlePubMedPMC
- 3. Scully T, Ettela A, LeRoith D, Gallagher EJ. Obesity, type 2 diabetes, and cancer risk. Front Oncol 2021;10:615375.ArticlePubMedPMC
- 4. Lega IC, Lipscombe LL. Review: diabetes, obesity, and cancerpathophysiology and clinical implications. Endocr Rev 2020;41:bnz014.ArticlePubMedPDF
- 5. Azrad M, Blair CK, Rock CL, Sedjo RL, Wolin KY, Demark-Wahnefried W. Adult weight gain accelerates the onset of breast cancer. Breast Cancer Res Treat 2019;176:649-56.ArticlePubMedPMCPDF
- 6. Pearson-Stuttard J, Zhou B, Kontis V, Bentham J, Gunter MJ, Ezzati M. Worldwide burden of cancer attributable to diabetes and high body-mass index: a comparative risk assessment. Lancet Diabetes Endocrinol 2018;6:e6-15.ArticlePMC
- 7. Kahn CR, Wang G, Lee KY. Altered adipose tissue and adipocyte function in the pathogenesis of metabolic syndrome. J Clin Invest 2019;129:3990-4000.ArticlePubMedPMC
- 8. Stern JH, Rutkowski JM, Scherer PE. Adiponectin, leptin, and fatty acids in the maintenance of metabolic homeostasis through adipose tissue crosstalk. Cell Metab 2016;23:770-84.ArticlePubMedPMC
- 9. Divella R, De Luca R, Abbate I, Naglieri E, Daniele A. Obesity and cancer: the role of adipose tissue and adipo-cytokines-induced chronic inflammation. J Cancer 2016;7:2346-59.ArticlePubMedPMC
- 10. Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med 2011;17:1498-503.ArticlePubMedPMCPDF
- 11. Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 2010;140:197-208.ArticlePubMedPMC
- 12. Hoy AJ, Balaban S, Saunders DN. Adipocyte-tumor cell metabolic crosstalk in breast cancer. Trends Mol Med 2017;23:381-92.ArticlePubMed
- 13. Cozzo AJ, Fuller AM, Makowski L. Contribution of adipose tissue to development of cancer. Compr Physiol 2017;8:237-82.ArticlePubMedPMCPDF
- 14. Samuel SM, Varghese E, Varghese S, Busselberg D. Challenges and perspectives in the treatment of diabetes associated breast cancer. Cancer Treat Rev 2018;70:98-111.ArticlePubMed
- 15. Garcia-Jimenez C, Garcia-Martinez JM, Chocarro-Calvo A, De la Vieja A. A new link between diabetes and cancer: enhanced WNT/β-catenin signaling by high glucose. J Mol Endocrinol 2013;52:R51-66.ArticlePubMed
- 16. Gallagher EJ, LeRoith D. The proliferating role of insulin and insulin-like growth factors in cancer. Trends Endocrinol Metab 2010;21:610-8.ArticlePubMedPMC
- 17. Novosyadlyy R, Lann DE, Vijayakumar A, Rowzee A, Lazzarino DA, Fierz Y, et al. Insulin-mediated acceleration of breast cancer development and progression in a nonobese model of type 2 diabetes. Cancer Res 2010;70:741-51.ArticlePubMedPMCPDF
- 18. Nelson ER, Wardell SE, Jasper JS, Park S, Suchindran S, Howe MK, et al. 27-Hydroxycholesterol links hypercholesterolemia and breast cancer pathophysiology. Science 2013;342:1094-8.ArticlePubMedPMC
- 19. Gallagher EJ, Fierz Y, Vijayakumar A, Haddad N, Yakar S, Le-Roith D. Inhibiting PI3K reduces mammary tumor growth and induces hyperglycemia in a mouse model of insulin resistance and hyperinsulinemia. Oncogene 2012;31:3213-22.ArticlePubMedPDF
- 20. Lengyel E, Makowski L, DiGiovanni J, Kolonin MG. Cancer as a matter of fat: the crosstalk between adipose tissue and tumors. Trends Cancer 2018;4:374-84.ArticlePubMedPMC
- 21. Park J, Euhus DM, Scherer PE. Paracrine and endocrine effects of adipose tissue on cancer development and progression. Endocr Rev 2011;32:550-70.ArticlePubMedPMC
- 22. Balaban S, Shearer RF, Lee LS, van Geldermalsen M, Schreuder M, Shtein HC, et al. Adipocyte lipolysis links obesity to breast cancer growth: adipocyte-derived fatty acids drive breast cancer cell proliferation and migration. Cancer Metab 2017;5:1.ArticlePubMedPMCPDF
- 23. Ouchi N, Parker JL, Lugus JJ, Walsh K. Adipokines in inflammation and metabolic disease. Nat Rev Immunol 2011;11:85-97.ArticlePubMedPMCPDF
- 24. Deng Y, Scherer PE. Adipokines as novel biomarkers and regulators of the metabolic syndrome. Ann N Y Acad Sci 2010;1212:E1-19.Article
- 25. Lehr S, Hartwig S, Sell H. Adipokines: a treasure trove for the discovery of biomarkers for metabolic disorders. Proteomics Clin Appl 2012;6:91-101.ArticlePubMedPDF
- 26. Kusminski CM, Bickel PE, Scherer PE. Targeting adipose tissue in the treatment of obesity-associated diabetes. Nat Rev Drug Discov 2016;15:639-60.ArticlePubMedPDF
- 27. Guaita-Esteruelas S, Bosquet A, Saavedra P, Guma J, Girona J, Lam EW, et al. Exogenous FABP4 increases breast cancer cell proliferation and activates the expression of fatty acid transport proteins. Mol Carcinog 2017;56:208-17.ArticlePubMedPDF
- 28. Hu MB, Xu H, Zhu WH, Bai PD, Hu JM, Yang T, et al. High-fat diet-induced adipokine and cytokine alterations promote the progression of prostate cancer in vivo and in vitro. Oncol Lett 2018;15:1607-15.ArticlePubMed
- 29. Peng C, Sun Z, Li O, Guo C, Yi W, Tan Z, et al. Leptin stimulates the epithelial-mesenchymal transition and pro-angiogenic capability of cholangiocarcinoma cells through the miR-122/PKM2 axis. Int J Oncol 2019;55:298-308.ArticlePubMed
- 30. Zhao C, Wu M, Zeng N, Xiong M, Hu W, Lv W, et al. Cancer-associated adipocytes: emerging supporters in breast cancer. J Exp Clin Cancer Res 2020;39:156.ArticlePubMedPMCPDF
- 31. Wang WJ, Lai HY, Zhang F, Shen WJ, Chu PY, Liang HY, et al. MCL1 participates in leptin-promoted mitochondrial fusion and contributes to drug resistance in gallbladder cancer. JCI Insight 2021;6:135438.Article
- 32. Zhao S, Kusminski CM, Scherer PE. Adiponectin, leptin and cardiovascular disorders. Circ Res 2021;128:136-49.ArticlePubMedPMC
- 33. Parida S, Siddharth S, Sharma D. Adiponectin, obesity, and cancer: clash of the bigwigs in health and disease. Int J Mol Sci 2019;20:2519.ArticlePubMedPMC
- 34. Scherer PE, Williams S, Fogliano M, Baldini G, Lodish HF. A novel serum protein similar to C1q, produced exclusively in adipocytes. J Biol Chem 1995;270:26746-9.ArticlePubMed
- 35. Berg AH, Combs TP, Scherer PE. ACRP30/adiponectin: an adipokine regulating glucose and lipid metabolism. Trends Endocrinol Metab 2002;13:84-9.ArticlePubMed
- 36. Dalamaga M, Diakopoulos KN, Mantzoros CS. The role of adiponectin in cancer: a review of current evidence. Endocr Rev 2012;33:547-94.ArticlePubMedPMCPDF
- 37. Jiang J, Fan Y, Zhang W, Shen Y, Liu T, Yao M, et al. Adiponectin suppresses human pancreatic cancer growth through attenuating the β-catenin signaling pathway. Int J Biol Sci 2019;15:253-64.ArticlePubMedPMC
- 38. Ye J, Jia J, Dong S, Zhang C, Yu S, Li L, et al. Circulating adiponectin levels and the risk of breast cancer: a meta-analysis. Eur J Cancer Prev 2014;23:158-65.PubMed
- 39. Miyoshi Y, Funahashi T, Kihara S, Taguchi T, Tamaki Y, Matsuzawa Y, et al. Association of serum adiponectin levels with breast cancer risk. Clin Cancer Res 2003;9:5699-704.PubMed
- 40. Landskroner-Eiger S, Qian B, Muise ES, Nawrocki AR, Berger JP, Fine EJ, et al. Proangiogenic contribution of adiponectin toward mammary tumor growth in vivo. Clin Cancer Res 2009;15:3265-76.ArticlePubMedPMCPDF
- 41. Di Zazzo E, Polito R, Bartollino S, Nigro E, Porcile C, Bianco A, et al. Adiponectin as link factor between adipose tissue and cancer. Int J Mol Sci 2019;20:839.ArticlePubMedPMC
- 42. Taliaferro-Smith L, Nagalingam A, Zhong D, Zhou W, Saxena NK, Sharma D. LKB1 is required for adiponectin-mediated modulation of AMPK-S6K axis and inhibition of migration and invasion of breast cancer cells. Oncogene 2009;28:2621-33.ArticlePubMedPMCPDF
- 43. Ouchi N, Kobayashi H, Kihara S, Kumada M, Sato K, Inoue T, et al. Adiponectin stimulates angiogenesis by promoting cross-talk between AMP-activated protein kinase and Akt signaling in endothelial cells. J Biol Chem 2004;279:1304-9.ArticlePubMed
- 44. Ouchi N, Kihara S, Arita Y, Okamoto Y, Maeda K, Kuriyama H, et al. Adiponectin, an adipocyte-derived plasma protein, inhibits endothelial NF-kappaB signaling through a cAMP-dependent pathway. Circulation 2000;102:1296-301.ArticlePubMed
- 45. Kim AY, Lee YS, Kim KH, Lee JH, Lee HK, Jang SH, et al. Adiponectin represses colon cancer cell proliferation via AdipoR1- and -R2-mediated AMPK activation. Mol Endocrinol 2010;24:1441-52.ArticlePubMedPMC
- 46. Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature 1994;372:425-32.ArticlePubMedPDF
- 47. Ray A, Cleary MP. The potential role of leptin in tumor invasion and metastasis. Cytokine Growth Factor Rev 2017;38:80-97.ArticlePubMedPMC
- 48. Yom CK, Lee KM, Han W, Kim SW, Kim HS, Moon BI, et al. Leptin as a potential target for estrogen receptor-positive breast cancer. J Breast Cancer 2013;16:138-45.ArticlePubMedPMC
- 49. Lipsey CC, Harbuzariu A, Robey RW, Huff LM, Gottesman MM, Gonzalez-Perez RR. Leptin signaling affects survival and chemoresistance of estrogen receptor negative breast cancer. Int J Mol Sci 2020;21:3794.ArticlePubMedPMC
- 50. Park J, Scherer PE. Leptin and cancer: from cancer stem cells to metastasis. Endocr Relat Cancer 2011;18:C25-9.ArticlePubMedPMC
- 51. Roder PV, Wu B, Liu Y, Han W. Pancreatic regulation of glucose homeostasis. Exp Mol Med 2016;48:e219.ArticlePubMedPMCPDF
- 52. Orgel E, Mittelman SD. The links between insulin resistance, diabetes, and cancer. Curr Diab Rep 2013;13:213-22.ArticlePubMedPMCPDF
- 53. de Luca C, Olefsky JM. Inflammation and insulin resistance. FEBS Lett 2008;582:97-105.ArticlePubMedPDF
- 54. Sriuranpong V, Park JI, Amornphimoltham P, Patel V, Nelkin BD, Gutkind JS. Epidermal growth factor receptor-independent constitutive activation of STAT3 in head and neck squamous cell carcinoma is mediated by the autocrine/paracrine stimulation of the interleukin 6/gp130 cytokine system. Cancer Res 2003;63:2948-56.PubMed
- 55. Payne AH, Hales DB. Overview of steroidogenic enzymes in the pathway from cholesterol to active steroid hormones. Endocr Rev 2004;25:947-70.ArticlePubMed
- 56. Park J, Morley TS, Kim M, Clegg DJ, Scherer PE. Obesity and cancer: mechanisms underlying tumour progression and recurrence. Nat Rev Endocrinol 2014;10:455-65.ArticlePubMedPMCPDF
- 57. Bulun SE, Chen D, Moy I, Brooks DC, Zhao H. Aromatase, breast cancer and obesity: a complex interaction. Trends Endocrinol Metab 2012;23:83-9.ArticlePubMed
- 58. Mukhopadhyay KD, Liu Z, Bandyopadhyay A, Kirma NB, Tekmal RR, Wang S, et al. Aromatase expression increases the survival and malignancy of estrogen receptor positive breast cancer cells. PLoS One 2015;10:e0121136.ArticlePubMedPMC
- 59. Chen D, Reierstad S, Fang F, Bulun SE. JunD and JunB integrate prostaglandin E2 activation of breast cancer-associated proximal aromatase promoters. Mol Endocrinol 2011;25:767-75.ArticlePubMedPMC
- 60. Chen D, Reierstad S, Lin Z, Lu M, Brooks C, Li N, et al. Prostaglandin E(2) induces breast cancer related aromatase promoters via activation of p38 and c-Jun NH(2)-terminal kinase in adipose fibroblasts. Cancer Res 2007;67:8914-22.ArticlePubMedPDF
- 61. Liang J, Shang Y. Estrogen and cancer. Annu Rev Physiol 2013;75:225-40.ArticlePubMed
- 62. Jensen EV, Cheng G, Palmieri C, Saji S, Makela S, Van Noorden S, et al. Estrogen receptors and proliferation markers in primary and recurrent breast cancer. Proc Natl Acad Sci U S A 2001;98:15197-202.ArticlePubMedPMC
- 63. Ungefroren H, Sebens S, Seidl D, Lehnert H, Hass R. Interaction of tumor cells with the microenvironment. Cell Commun Signal 2011;9:18.ArticlePubMedPMC
- 64. Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009;139:891-906.ArticlePubMedPMC
- 65. Wang M, Zhao J, Zhang L, Wei F, Lian Y, Wu Y, et al. Role of tumor microenvironment in tumorigenesis. J Cancer 2017;8:761-73.ArticlePubMedPMC
- 66. Henke E, Nandigama R, Ergun S. Extracellular matrix in the tumor microenvironment and its impact on cancer therapy. Front Mol Biosci 2020;6:160.ArticlePubMedPMC
- 67. Romero-Lopez M, Trinh AL, Sobrino A, Hatch MM, Keating MT, Fimbres C, et al. Recapitulating the human tumor microenvironment: colon tumor-derived extracellular matrix promotes angiogenesis and tumor cell growth. Biomaterials 2017;116:118-29.ArticlePubMed
- 68. Ulrich TA, de Juan Pardo EM, Kumar S. The mechanical rigidity of the extracellular matrix regulates the structure, motility, and proliferation of glioma cells. Cancer Res 2009;69:4167-74.ArticlePubMedPMCPDF
- 69. Ferruzzi J, Sun M, Gkousioudi A, Pilvar A, Roblyer D, Zhang Y, et al. Compressive remodeling alters fluid transport properties of collagen networks: implications for tumor growth. Sci Rep 2019;9:17151.ArticlePubMedPMCPDF
- 70. Le CC, Bennasroune A, Langlois B, Salesse S, BoulagnonRombi C, Morjani H, et al. Functional interplay between collagen network and cell behavior within tumor microenvironment in colorectal cancer. Front Oncol 2020;10:527.ArticlePubMedPMC
- 71. Stanisavljevic J, Loubat-Casanovas J, Herrera M, Luque T, Pena R, Lluch A, et al. Snail1-expressing fibroblasts in the tumor microenvironment display mechanical properties that support metastasis. Cancer Res 2015;75:284-95.ArticlePubMedPDF
- 72. Muz B, de la Puente P, Azab F, Azab AK. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia (Auckl) 2015;3:83-92.ArticlePubMedPMC
- 73. Jing X, Yang F, Shao C, Wei K, Xie M, Shen H, et al. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol Cancer 2019;18:157.ArticlePubMedPMCPDF
- 74. Jiang Z, Hsu JL, Li Y, Hortobagyi GN, Hung MC. Cancer cell metabolism bolsters immunotherapy resistance by promoting an immunosuppressive tumor microenvironment. Front Oncol 2020;10:1197.ArticlePubMedPMC
- 75. Kheshtchin N, Arab S, Ajami M, Mirzaei R, Ashourpour M, Mousavi N, et al. Inhibition of HIF-1α enhances anti-tumor effects of dendritic cell-based vaccination in a mouse model of breast cancer. Cancer Immunol Immunother 2016;65:1159-67.ArticlePubMedPDF
- 76. Ye LY, Chen W, Bai XL, Xu XY, Zhang Q, Xia XF, et al. Hypoxia-induced epithelial-to-mesenchymal transition in hepatocellular carcinoma induces an immunosuppressive tumor microenvironment to promote metastasis. Cancer Res 2016;76:818-30.ArticlePubMedPDF
- 77. Gong C, Yu X, Zhang W, Han L, Wang R, Wang Y, et al. Regulating the immunosuppressive tumor microenvironment to enhance breast cancer immunotherapy using pH-responsive hybrid membrane-coated nanoparticles. J Nanobiotechnology 2021;19:58.ArticlePubMedPMCPDF
- 78. Wu Q, Li B, Li Z, Li J, Sun S, Sun S. Cancer-associated adipocytes: key players in breast cancer progression. J Hematol Oncol 2019;12:95.ArticlePubMedPMCPDF
- 79. Kulik G, Weber MJ. Akt-dependent and -independent survival signaling pathways utilized by insulin-like growth factor I. Mol Cell Biol 1998;18:6711-8.ArticlePubMedPMCPDF
- 80. Simpson A, Petnga W, Macaulay VM, Weyer-Czernilofsky U, Bogenrieder T. Insulin-like growth factor (IGF) pathway targeting in cancer: role of the IGF axis and opportunities for future combination studies. Target Oncol 2017;12:571-97.ArticlePubMedPMCPDF
- 81. Huang YK, Kang WM, Ma ZQ, Liu YQ, Zhou L, Yu JC. NU-CKS1 promotes gastric cancer cell aggressiveness by upregulating IGF-1R and subsequently activating the PI3K/Akt/mTOR signaling pathway. Carcinogenesis 2019;40:370-9.ArticlePubMed
- 82. Fruhbeck G. Intracellular signalling pathways activated by leptin. Biochem J 2006;393(Pt 1):7-20.ArticlePubMedPDF
- 83. Sanchez-Jimenez F, Perez-Perez A, de la Cruz-Merino L, Sanchez-Margalet V. Obesity and breast cancer: role of leptin. Front Oncol 2019;9:596.PubMedPMC
- 84. Saxena NK, Taliaferro-Smith L, Knight BB, Merlin D, Anania FA, O’Regan RM, et al. Bidirectional crosstalk between leptin and insulin-like growth factor-I signaling promotes invasion and migration of breast cancer cells via transactivation of epidermal growth factor receptor. Cancer Res 2008;68:9712-22.ArticlePubMedPMCPDF
- 85. Chandrasekar B, Boylston WH, Venkatachalam K, Webster NJ, Prabhu SD, Valente AJ. Adiponectin blocks interleukin18-mediated endothelial cell death via APPL1-dependent AMP-activated protein kinase (AMPK) activation and IKK/NF-kappaB/PTEN suppression. J Biol Chem 2008;283:24889-98.PubMedPMC
- 86. Chocarro-Calvo A, Garcia-Martinez JM, Ardila-Gonzalez S, De la Vieja A, Garcia-Jimenez C. Glucose-induced β-catenin acetylation enhances Wnt signaling in cancer. Mol Cell 2013;49:474-86.ArticlePubMed
- 87. Dirat B, Bochet L, Dabek M, Daviaud D, Dauvillier S, Majed B, et al. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res 2011;71:2455-65.ArticlePubMedPDF
- 88. Liberti MV, Locasale JW. The warburg effect: how does it benefit cancer cells? Trends Biochem Sci 2016;41:211-8.ArticlePubMedPMC
- 89. Xu Y, Tan M, Tian X, Zhang J, Zhang J, Chen J, et al. Leptin receptor mediates the proliferation and glucose metabolism of pancreatic cancer cells via AKT pathway activation. Mol Med Rep 2020;21:945-52.ArticlePubMed
- 90. Wei L, Li K, Pang X, Guo B, Su M, Huang Y, et al. Leptin promotes epithelial-mesenchymal transition of breast cancer via the upregulation of pyruvate kinase M2. J Exp Clin Cancer Res 2016;35:166.ArticlePubMedPMCPDF
- 91. Pham DV, Park PH. Tumor metabolic reprogramming by adipokines as a critical driver of obesity-associated cancer progression. Int J Mol Sci 2021;22:1444.ArticlePubMedPMC
- 92. Huang J, Duran A, Reina-Campos M, Valencia T, Castilla EA, Muller TD, et al. Adipocyte p62/SQSTM1 suppresses tumorigenesis through opposite regulations of metabolism in adipose tissue and tumor. Cancer Cell 2018;33:770-84.ArticlePubMedPMC
- 93. Cerf ME. Beta cell dysfunction and insulin resistance. Front Endocrinol (Lausanne) 2013;4:37.ArticlePubMedPMC
- 94. Garg SK, Maurer H, Reed K, Selagamsetty R. Diabetes and cancer: two diseases with obesity as a common risk factor. Diabetes Obes Metab 2014;16:97-110.ArticlePubMedPDF
- 95. Suh S, Kim KW. Diabetes and cancer: cancer should be screened in routine diabetes assessment. Diabetes Metab J 2019;43:733-43.ArticlePubMedPMCPDF
- 96. Libby G, Donnelly LA, Donnan PT, Alessi DR, Morris AD, Evans JM. New users of metformin are at low risk of incident cancer: a cohort study among people with type 2 diabetes. Diabetes Care 2009;32:1620-5.PubMedPMC
- 97. Bodmer M, Meier C, Krahenbuhl S, Jick SS, Meier CR. Longterm metformin use is associated with decreased risk of breast cancer. Diabetes Care 2010;33:1304-8.ArticlePubMedPMCPDF
- 98. Lee JH, Kim TI, Jeon SM, Hong SP, Cheon JH, Kim WH. The effects of metformin on the survival of colorectal cancer patients with diabetes mellitus. Int J Cancer 2012;131:752-9.ArticlePubMed
- 99. Goodwin PJ, Ennis M, Bahl M, Fantus IG, Pritchard KI, Trudeau ME, et al. High insulin levels in newly diagnosed breast cancer patients reflect underlying insulin resistance and are associated with components of the insulin resistance syndrome. Breast Cancer Res Treat 2009;114:517-25.ArticlePubMedPDF
- 100. Boyle P, Boniol M, Koechlin A, Robertson C, Valentini F, Coppens K, et al. Diabetes and breast cancer risk: a meta-analysis. Br J Cancer 2012;107:1608-17.ArticlePubMedPMCPDF
- 101. Ben Q, Xu M, Ning X, Liu J, Hong S, Huang W, et al. Diabetes mellitus and risk of pancreatic cancer: a meta-analysis of cohort studies. Eur J Cancer 2011;47:1928-37.ArticlePubMed
- 102. Lambe M, Wigertz A, Garmo H, Walldius G, Jungner I, Hammar N. Impaired glucose metabolism and diabetes and the risk of breast, endometrial, and ovarian cancer. Cancer Causes Control 2011;22:1163-71.ArticlePubMedPDF
- 103. Hopkins BD, Goncalves MD, Cantley LC. Insulin-PI3K signalling: an evolutionarily insulated metabolic driver of cancer. Nat Rev Endocrinol 2020;16:276-83.ArticlePubMedPMCPDF
- 104. Sundaram S, Johnson AR, Makowski L. Obesity, metabolism and the microenvironment: links to cancer. J Carcinog 2013;12:19.ArticlePubMedPMC
- 105. Desbois-Mouthon C, Cadoret A, Blivet-Van Eggelpoel MJ, Bertrand F, Cherqui G, Perret C, et al. Insulin and IGF-1 stimulate the beta-catenin pathway through two signalling cascades involving GSK-3beta inhibition and Ras activation. Oncogene 2001;20:252-9.ArticlePubMedPDF
- 106. Easwaran V, Lee SH, Inge L, Guo L, Goldbeck C, Garrett E, et al. Beta-catenin regulates vascular endothelial growth factor expression in colon cancer. Cancer Res 2003;63:3145-53.PubMed
- 107. Zhang Y, Moerkens M, Ramaiahgari S, de Bont H, Price L, Meerman J, et al. Elevated insulin-like growth factor 1 receptor signaling induces antiestrogen resistance through the MAPK/ERK and PI3K/Akt signaling routes. Breast Cancer Res 2011;13:R52.ArticlePubMedPMCPDF
- 108. Goldberg IJ. Clinical review 124: diabetic dyslipidemia: causes and consequences. J Clin Endocrinol Metab 2001;86:965-71.PubMed
- 109. He S, Nelson ER. 27-Hydroxycholesterol, an endogenous selective estrogen receptor modulator. Maturitas 2017;104:29-35.ArticlePubMedPMC
- 110. Liu L, Li MY, Xing Y, Wang XY, Wang Y. The oncogenic roles of 27-hydroxycholesterol in glioblastoma. Oncol Lett 2019;18:3623-9.ArticlePubMedPMC
- 111. Kimbung S, Chang CY, Bendahl PO, Dubois L, Thompson JW, McDonnell DP, et al. Impact of 27-hydroxylase (CYP27A1) and 27-hydroxycholesterol in breast cancer. Endocr Relat Cancer 2017;24:339-49.ArticlePubMed
- 112. Kimbung S, Inasu M, Stalhammar T, Nodin B, Elebro K, Tryggvadottir H, et al. CYP27A1 expression is associated with risk of late lethal estrogen receptor-positive breast cancer in postmenopausal patients. Breast Cancer Res 2020;22:123.ArticlePubMedPMCPDF
- 113. Guillemot-Legris O, Mutemberezi V, Cani PD, Muccioli GG. Obesity is associated with changes in oxysterol metabolism and levels in mice liver, hypothalamus, adipose tissue and plasma. Sci Rep 2016;6:19694.ArticlePubMedPMCPDF
- 114. Griffin ME, Marcucci MJ, Cline GW, Bell K, Barucci N, Lee D, et al. Free fatty acid-induced insulin resistance is associated with activation of protein kinase C theta and alterations in the insulin signaling cascade. Diabetes 1999;48:1270-4.ArticlePubMedPDF
- 115. Poornima IG, Parikh P, Shannon RP. Diabetic cardiomyopathy: the search for a unifying hypothesis. Circ Res 2006;98:596-605.PubMed
- 116. Beloribi-Djefaflia S, Vasseur S, Guillaumond F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016;5:e189.ArticlePubMedPMCPDF
- 117. Mollinedo F, Gajate C. Lipid rafts as signaling hubs in cancer cell survival/death and invasion: implications in tumor progression and therapy: thematic review series: biology of lipid rafts. J Lipid Res 2020;61:611-35.PubMedPMC
- 118. Liu YY, Han TY, Giuliano AE, Cabot MC. Ceramide glycosylation potentiates cellular multidrug resistance. FASEB J 2001;15:719-30.ArticlePubMedPDF
- 119. He X, Esteva FJ, Ensor J, Hortobagyi GN, Lee MH, Yeung SC. Metformin and thiazolidinediones are associated with improved breast cancer-specific survival of diabetic women with HER2+ breast cancer. Ann Oncol 2012;23:1771-80.ArticlePubMed
- 120. Fong D, Christensen CT, Chan MM. Targeting cancer stem cells with repurposed drugs to improve current therapies. Recent Pat Anticancer Drug Discov 2021;16:136-60.ArticlePubMedPDF
- 121. Behrouzi B, Zokaasadi M, Mohagheghi MA, Emami AH, Sadighi S. The effect of metformin on survival outcomes of nonmetastatic breast cancer patients with type 2 diabetes. Asian Pac J Cancer Prev 2021;22:611-6.ArticlePubMedPMC
- 122. Saraei P, Asadi I, Kakar MA, Moradi-Kor N. The beneficial effects of metformin on cancer prevention and therapy: a comprehensive review of recent advances. Cancer Manag Res 2019;11:3295-313.PubMedPMC
- 123. Zhao B, Luo J, Yu T, Zhou L, Lv H, Shang P. Anticancer mechanisms of metformin: a review of the current evidence. Life Sci 2020;254:117717.ArticlePubMed
- 124. Vancura A, Bu P, Bhagwat M, Zeng J, Vancurova I. Metformin as an anticancer agent. Trends Pharmacol Sci 2018;39:867-78.ArticlePubMedPMC
- 125. Chang CH, Lin JW, Wu LC, Lai MS, Chuang LM, Chan KA. Association of thiazolidinediones with liver cancer and colorectal cancer in type 2 diabetes mellitus. Hepatology 2012;55:1462-72.ArticlePubMed
- 126. Arvind A, Memel ZN, Philpotts LL, Zheng H, Corey KE, Simon TG. Thiazolidinediones, alpha-glucosidase inhibitors, meglitinides, sulfonylureas, and hepatocellular carcinoma risk: a meta-analysis. Metabolism 2021;120:154780.ArticlePubMedPMC
- 127. Song R. Mechanism of metformin: a tale of two sites. Diabetes Care 2016;39:187-9.ArticlePubMedPDF
- 128. Lei Y, Yi Y, Liu Y, Liu X, Keller ET, Qian CN, et al. Metformin targets multiple signaling pathways in cancer. Chin J Cancer 2017;36:17.ArticlePubMedPMCPDF
- 129. He K, Hu H, Ye S, Wang H, Cui R, Yi L. The effect of metformin therapy on incidence and prognosis in prostate cancer: a systematic review and meta-analysis. Sci Rep 2019;9:2218.ArticlePubMedPMCPDF
- 130. Tsilidis KK, Capothanassi D, Allen NE, Rizos EC, Lopez DS, van Veldhoven K, et al. Metformin does not affect cancer risk: a cohort study in the U.K. Clinical Practice Research Datalink analyzed like an intention-to-treat trial. Diabetes Care 2014;37:2522-32.ArticlePubMedPDF
- 131. Stevens RJ, Ali R, Bankhead CR, Bethel MA, Cairns BJ, Camisasca RP, et al. Cancer outcomes and all-cause mortality in adults allocated to metformin: systematic review and collaborative meta-analysis of randomised clinical trials. Diabetologia 2012;55:2593-603.ArticlePubMedPDF
- 132. Chiarelli F, Di Marzio D. Peroxisome proliferator-activated receptor-gamma agonists and diabetes: current evidence and future perspectives. Vasc Health Risk Manag 2008;4:297-304.PubMedPMC
- 133. Blanquicett C, Roman J, Hart CM. Thiazolidinediones as anticancer agents. Cancer Ther 2008;6(A):25-34.PubMedPMC
- 134. Monami M, Lamanna C, Marchionni N, Mannucci E. Rosiglitazone and risk of cancer: a meta-analysis of randomized clinical trials. Diabetes Care 2008;31:1455-60.PubMedPMC
- 135. Koro C, Barrett S, Qizilbash N. Cancer risks in thiazolidinedione users compared to other anti-diabetic agents. Pharmacoepidemiol Drug Saf 2007;16:485-92.ArticlePubMed
- 136. Du R, Lin L, Cheng D, Xu Y, Xu M, Chen Y, et al. Thiazolidinedione therapy and breast cancer risk in diabetic women: a systematic review and meta-analysis. Diabetes Metab Res Rev 2018;34:e2961.ArticlePDF
REFERENCES
Figure & Data
References
Citations
- Triglyceride‐glucose index (TyG index) and endometrial carcinoma risk: A retrospective cohort study
Haimeng Shi, Feifei Guo, Kang Zheng, Rong Li, Huaijun Zhou
International Journal of Gynecology & Obstetrics.2024; 164(1): 298. CrossRef - Association between total cholesterol levels and all-cause mortality among newly diagnosed patients with cancer
Seohyun Kim, Gyuri Kim, So Hyun Cho, Rosa Oh, Ji Yoon Kim, You-Bin Lee, Sang-Man Jin, Kyu Yeon Hur, Jae Hyeon Kim
Scientific Reports.2024;[Epub] CrossRef - The Role of Adipokines in Tumor Progression and Its Association with Obesity
Jae Won Kim, Jun Hyeok Kim, Yoon Jae Lee
Biomedicines.2024; 12(1): 97. CrossRef - Triglyceride-glucose index is a risk factor for breast cancer in China: a cross-sectional study
Jinghua Zhang, Binbin Yin, Ya Xi, Yongying Bai
Lipids in Health and Disease.2024;[Epub] CrossRef - Insulin Resistance: The Increased Risk of Cancers
Leszek Szablewski
Current Oncology.2024; 31(2): 998. CrossRef - Epigenetic modifications in obesity‐associated diseases
Yiqian Long, Chao Mao, Shuang Liu, Yongguang Tao, Desheng Xiao
MedComm.2024;[Epub] CrossRef - The Juncture of Diabetes, Cancer, and Observational Population-based Studies: Novel Insights From Canadian Provincial Health Records
Terra Arnason, Kerry Mansell
Canadian Journal of Diabetes.2024;[Epub] CrossRef - The impact of chemotherapy on adipose tissue remodeling: The molecular players involved in this tissue wasting
Samuel Barbosa, Mafalda Barbosa Pedrosa, Rita Ferreira, Daniel Moreira-Gonçalves, Lúcio Lara Santos
Biochimie.2024; 223: 1. CrossRef - Epidemiological and transcriptome data identify shared gene signatures and immune cell infiltration in type 2 diabetes and non-small cell lung cancer
Qian Yuan, Long Li, Liu-shun Wang, Shi-gui Xing
Diabetology & Metabolic Syndrome.2024;[Epub] CrossRef - THE INFLUENCE OF DIABETES-ASSOCIATED FACTORS OF ONCOGENESIS ON THE RISK OF BREAST AND ENDOMETRIAL CANCER AND ON THE SURVIVAL OF WOMEN WITH THIS CANCER
Tamara S. Vatseba, Liubov K. Sokolova, Vasyl Ye. Neyko, Valentyna V. Dzvonkovska, Oksana V. Muravlova, Volodymyr V. Derpak
Clinical and Preventive Medicine.2024; (2): 99. CrossRef - Associations between diabetes and cancer: A 10-year national population-based retrospective cohort study
Heléna Safadi, Ágnes Balogh, Judit Lám, Attila Nagy, Éva Belicza
Diabetes Research and Clinical Practice.2024; 211: 111665. CrossRef - Colorectal Cancer: Epidemiology, Risk Factors, and Prevention
Gholamreza Roshandel, Fatemeh Ghasemi-Kebria, Reza Malekzadeh
Cancers.2024; 16(8): 1530. CrossRef - Metabolic Alteration Bridging the Prediabetic State and Colorectal Cancer
Antonino Colloca, Isabella Donisi, Camilla Anastasio, Maria Luisa Balestrieri, Nunzia D’Onofrio
Cells.2024; 13(8): 663. CrossRef - Ultra‐processed food consumption and the risk of pancreatic cancer in the Prostate, Lung, Colorectal and Ovarian Cancer Screening Trial
Guo‐Chao Zhong, Qian Zhu, Dong Cai, Jie‐Jun Hu, Xin Dai, Jian‐Ping Gong, Wei‐Ping Sun
International Journal of Cancer.2023; 152(5): 835. CrossRef - Advances in “adiponcosis”: Insights in the inner mechanisms at the base of adipose and tumour tissues interplay
Cristina Pagano, Erika di Zazzo, Giorgio Avilia, Beatrice Savarese, Giovanna Navarra, Maria Chiara Proto, Donatella Fiore, Monica Rienzo, Patrizia Gazzerro, Chiara Laezza, Maurizio Bifulco
International Journal of Cancer.2023; 152(12): 2464. CrossRef - Metabolites as signalling molecules
Steven Andrew Baker, Jared Rutter
Nature Reviews Molecular Cell Biology.2023; 24(5): 355. CrossRef - Characterization of neuroendocrine regulation- and metabolism-associated molecular features and prognostic indicators with aid to clinical chemotherapy and immunotherapy of patients with pancreatic cancer
Biao Zhang, Qihang Yuan, Bolin Zhang, Shuang Li, Zhizhou Wang, Hangyu Liu, Fanyue Meng, Xu Chen, Dong Shang
Frontiers in Endocrinology.2023;[Epub] CrossRef - Concentration of Selected Adipokines and Factors Regulating Carbohydrate Metabolism in Patients with Head and Neck Cancer in Respect to Their Body Mass Index
Jarosław Nuszkiewicz, Jolanta Czuczejko, Wiktor Dróżdż, Alina Woźniak, Bogdan Małkowski, Karolina Szewczyk-Golec
International Journal of Molecular Sciences.2023; 24(4): 3283. CrossRef - Hormonal Gut–Brain Signaling for the Treatment of Obesity
Eun Roh, Kyung Mook Choi
International Journal of Molecular Sciences.2023; 24(4): 3384. CrossRef - A novel five-gene metabolism-related risk signature for predicting prognosis and immune infiltration in endometrial cancer: A TCGA data mining
Huaqing Huang, Xintong Cai, Jiexiang Lin, Qiaoling Wu, Kailin Zhang, Yibin Lin, Bin Liu, Jie Lin
Computers in Biology and Medicine.2023; 155: 106632. CrossRef - Green Antimicrobials as Therapeutic Agents for Diabetic Foot Ulcers
Ines D. Teixeira, Eugenia Carvalho, Ermelindo C. Leal
Antibiotics.2023; 12(3): 467. CrossRef - Impact of abdominal obesity on the risk of glioma development in patients with diabetes: A nationwide population-based cohort study in Korea
Hyunji Sang, Yun Kyung Cho, Kyungdo Han, Eun Hee Koh, Sandar Tin Tin
PLOS ONE.2023; 18(3): e0283023. CrossRef - Relationship Between Physical Exercise and Cognitive Impairment Among Older Adults with Type 2 Diabetes: Chain Mediating Roles of Sleep Quality and Depression
Han Zhang, Yefan Zhang, Sen Sheng, Yang Xing, Zhongchen Mou, Yanqiu Zhang, Zhixue Shi, Zhenjie Yu, Qianqian Gao, Weiqin Cai, Qi Jing
Psychology Research and Behavior Management.2023; Volume 16: 817. CrossRef - The Mediterranean Lifestyle to Contrast Low-Grade Inflammation Behavior in Cancer
Rosa Divella, Graziella Marino, Stefania Infusino, Laura Lanotte, Gaia Gadaleta-Caldarola, Gennaro Gadaleta-Caldarola
Nutrients.2023; 15(7): 1667. CrossRef - Distribution of ABO blood groups and Rh factor in benign and malign thyroid nodules
Muzaffer Serdar DENİZ
Journal of Health Sciences and Medicine.2023; 6(2): 462. CrossRef - The Role of Selected Adipocytokines in Ovarian Cancer and Endometrial Cancer
Sebastian Stępień, Paweł Olczyk, Joanna Gola, Katarzyna Komosińska-Vassev, Aleksandra Mielczarek-Palacz
Cells.2023; 12(8): 1118. CrossRef - Coronary Revascularization in Patients With Cancer
Bala Pushparaji, Teodora Donisan, Dinu Valentin Balanescu, Jong Kun Park, Dominique J. Monlezun, Abdelrahman Ali, Ibrahim Halil Inanc, Jaime Caballero, Mehmet Cilingiroglu, Konstantinos Marmagkiolis, Cezar Iliescu
Current Treatment Options in Cardiovascular Medicine.2023; 25(6): 143. CrossRef - Hepatocellular carcinoma in a large cohort of type 2 diabetes patients
Carlo B. Giorda, Roberta Picariello, Barbara Tartaglino, Elisa Nada, Giuseppe Costa, Roberta Manti, Luca Monge, Roberto Gnavi
Diabetes Research and Clinical Practice.2023; 200: 110684. CrossRef - Prediction of Hypoglycemia in Diabetic Patients During Colonoscopy
Preparation
Xiaohua Lu, Lingqiao Xie, Wane Zhao, Chuangbiao Zhang, Xixi Luo, Yan Zhou
Experimental and Clinical Endocrinology & Diabetes.2023; 131(05): 274. CrossRef - Interplay of skeletal muscle and adipose tissue: sarcopenic obesity
Min Jeong Park, Kyung Mook Choi
Metabolism.2023; 144: 155577. CrossRef - PIK3CA mutation in endometriotic epithelial cells promotes viperin-dependent inflammatory response to insulin
Mike R. Wilson, Shannon Harkins, Jake J. Reske, Rebecca A. Siwicki, Marie Adams, Victoria L. Bae-Jump, Jose M. Teixeira, Ronald L. Chandler
Reproductive Biology and Endocrinology.2023;[Epub] CrossRef - The Function of MondoA and ChREBP Nutrient—Sensing Factors in Metabolic Disease
Byungyong Ahn
International Journal of Molecular Sciences.2023; 24(10): 8811. CrossRef - Mechanism of Cephalophyllum cephalus Intervention in Diabetes Based on Network Pharmacology and Molecular Docking Technology
志梁 范
Hans Journal of Medicinal Chemistry.2023; 11(02): 81. CrossRef - Development and validation of a prognostic model based on metabolic risk score to predict overall survival of endometrial cancer in Chinese patients
Xingchen Li, Xiao Yang, Yuan Cheng, Yangyang Dong, Jingyuan Wang, Jianliu Wang
Journal of Gynecologic Oncology.2023;[Epub] CrossRef - Nε-(1-Carboxymethyl)-L-lysine, an advanced glycation end product, exerts malignancy on chondrosarcoma via the activation of cancer stemness
Ting-Yu Chang, Kuo-Cheng Lan, Chia-Hung Wu, Meei-Ling Sheu, Rong-Sen Yang, Shing-Hwa Liu
Archives of Toxicology.2023; 97(8): 2231. CrossRef - The Link between Diabetes, Pancreatic Tumors, and miRNAs—New Players for Diagnosis and Therapy?
Małgorzata Kozłowska, Agnieszka Śliwińska
International Journal of Molecular Sciences.2023; 24(12): 10252. CrossRef - Obesity, diabetes, and cancer: epidemiology, pathophysiology, and potential interventions
Leonardo de Andrade Mesquita, Laura Fink Wayerbacher, Gilberto Schwartsmann, Fernando Gerchman
Archives of Endocrinology and Metabolism.2023;[Epub] CrossRef - Distinct Lipid Phenotype of Cancer-Associated Fibroblasts (CAFs) Isolated From Overweight/Obese Endometrial Cancer Patients as Assessed Using Raman Spectroscopy
Tze Hua Yeu, Intan Sofia Omar, S.F. Abdul Sani, Dharini Pathmanathan, Boon Tong Goh, Nithyialakshmi Ravindran, Ik Hui Teo, Yogeeta Gunasagran, Noor Azmi Mat Adenan, Ivy Chung, Amira Hajirah Abd Jamil
Applied Spectroscopy.2023; 77(7): 723. CrossRef - Global, Regional, and National Epidemiology of Diabetes in Children From 1990 to 2019
Kexin Zhang, Chengxia Kan, Fang Han, Jingwen Zhang, Chuanhua Ding, Zhentao Guo, Na Huang, Yang Zhang, Ningning Hou, Xiaodong Sun
JAMA Pediatrics.2023; 177(8): 837. CrossRef - The effects of physical exercise therapy on weight control: its regulation of adipocyte physiology and metabolic capacity
Hyun Jung Park, Sung Ja Rhie, Insop Shim
Journal of Exercise Rehabilitation.2023; 19(3): 141. CrossRef - The Ephrin tyrosine kinase a3 (EphA3) is a novel mediator of RAGE-prompted motility of breast cancer cells
Marianna Talia, Francesca Cirillo, Asia Spinelli, Azzurra Zicarelli, Domenica Scordamaglia, Lucia Muglia, Salvatore De Rosis, Damiano Cosimo Rigiracciolo, Gianfranco Filippelli, Ida Daniela Perrotta, Mariano Davoli, Rosanna De Rosa, Rachele Macirella, Elv
Journal of Experimental & Clinical Cancer Research.2023;[Epub] CrossRef - Signal detection in real‐world data and confirmation in clinical trials: Diabetes as a case study for a conversation worth having
Elad Sharon, Megan Othus, Zoe Eloise Quandt
Cancer.2023; 129(18): 2769. CrossRef - Ultra-Processed Food Consumption and Obesity in Korean Adults
Jee-Seon Shim, Kyoung Hwa Ha, Dae Jung Kim, Hyeon Chang Kim
Diabetes & Metabolism Journal.2023; 47(4): 547. CrossRef - Decoding the role of leptin and adiponectin in obesity-related gastrointestinal cancer
Vanda Marques, Fabiola Arella, Marta B. Afonso, André A. Santos, Cecília M.P. Rodrigues
Clinical Science.2023; 137(15): 1095. CrossRef - Diabetes and two kinds of primary tumors in a patient with thalassemia: a case report and literature review
Xiaoyan Yu, Yi Peng, Tingting Nie, Wenjia Sun, Yajuan Zhou
Frontiers in Oncology.2023;[Epub] CrossRef - Additive impact of diabetes and sarcopenia on all-cause and cardiovascular mortality: A longitudinal nationwide population-based study
Eyun Song, Soon Young Hwang, Min Jeong Park, Ahreum Jang, Kyeong Jin Kim, Ji Hee Yu, Nam Hoon Kim, Hye Jin Yoo, Ji A. Seo, Sin Gon Kim, Nan Hee Kim, Sei Hyun Baik, Kyung Mook Choi
Metabolism.2023; 148: 155678. CrossRef - Obesity and Colorectal Cancer
Jundeok Lee, Su Young Kim
The Korean Journal of Gastroenterology.2023; 82(2): 63. CrossRef - Cognitive Behavioural Therapies for Weight-Loss in Adults: A Scoping Review Protocol
Laura María Compañ-Gabucio, Diana Mancheño-Bañón, Laura Torres-Collado, Jesús Vioque, Manuela García-de-la-Hera
Healthcare.2023; 11(18): 2473. CrossRef - Saxagliptin, a selective dipeptidyl peptidase-4 inhibitor, alleviates somatic cell aneugenicity and clastogenicity in diabetic mice
Sabry M. Attia, Sheikh F. Ahmad, Ahmed Nadeem, Mohamed S.M. Attia, Mushtaq A. Ansari, Abdelkader E. Ashour, Norah A. Albekairi, Mohammed A. Al-Hamamah, Ali A. Alshamrani, Saleh A. Bakheet
Mutation Research/Genetic Toxicology and Environmental Mutagenesis.2023; 892: 503707. CrossRef - Mechanisms of Linggui Zhugan Decoction in the Treatment of Obesity Based on Network Pharmacology
Chunmei Liu, Li Zhang, Yubin Yang
Science of Advanced Materials.2023; 15(9): 1265. CrossRef - Beyond Body Size: Adiponectin as a Key Player in Obesity-Driven Cancers
Maurizio Capuozzo, Venere Celotto, Loris Landi, Francesco Ferrara, Francesco Sabbatino, Francesco Perri, Marco Cascella, Vincenza Granata, Mariachiara Santorsola, Alessandro Ottaiano
Nutrition and Cancer.2023; 75(10): 1848. CrossRef - Impact of sarcopenic obesity on post-hepatectomy bile leakage for hepatocellular carcinoma
Hikaru Hayashi, Akira Shimizu, Koji Kubota, Tsuyoshi Notake, Hitoshi Masuo, Takahiro Yoshizawa, Kiyotaka Hosoda, Hiroki Sakai, Koya Yasukawa, Yuji Soejima, Ezio Lanza
PLOS ONE.2023; 18(10): e0286353. CrossRef - Colorectal Cancer: From Risk Factors to Oncogenesis
Vlad Alexandru Ionescu, Gina Gheorghe, Nicolae Bacalbasa, Alexandru Laurentiu Chiotoroiu, Camelia Diaconu
Medicina.2023; 59(9): 1646. CrossRef - The impact of poor metabolic health on aggressive breast cancer: adipose tissue and tumor metabolism
Barbara Mensah Sankofi, Estefania Valencia-Rincón, Malika Sekhri, Adriana L. Ponton-Almodovar, Jamie J. Bernard, Elizabeth A. Wellberg
Frontiers in Endocrinology.2023;[Epub] CrossRef - Association of nonalcoholic fatty liver disease and growth hormone deficiency: a systematic review and meta-analysis
Tingting Kong, Yunpeng Gu, Lei Sun, Run Zhou, Jie Li, Junping Shi
Endocrine Journal.2023; 70(10): 959. CrossRef - Impact of Caloric Restriction in Breast Cancer Patients Treated with Neoadjuvant Chemotherapy: A Prospective Case Control Study
Isabella Castellano, Francesco Gallo, Paola Durelli, Taira Monge, Maurizio Fadda, Jasna Metovic, Paola Cassoni, Fulvio Borella, Carlo Raucci, Monica Menischetti, Alessandra Beano, Giuseppe Migliaretti, Concetta Finocchiaro
Nutrients.2023; 15(21): 4677. CrossRef - Metabolomic and lipidomic studies on the intervention of taurochenodeoxycholic acid in mice with hyperlipidemia
Na Cui, Wensen Zhang, Fazhi Su, Zhihong Zhang, Biao Li, Donghui Peng, Yanping Sun, Yuanning Zeng, Bingyou Yang, Haixue Kuang, Qiuhong Wang
Frontiers in Pharmacology.2023;[Epub] CrossRef - Is MG53 a potential therapeutic target for cancer?
Yunyu Du, Tieying Li, Muqing Yi
Frontiers in Endocrinology.2023;[Epub] CrossRef - Blood Biomarkers Panels for Screening of Colorectal Cancer and Adenoma on a Machine Learning-Assisted Detection Platform
Hui Wang, Zhiwei Zhou, Haijun Li, Weiguang Xiang, Yilin Lan, Xiaowen Dou, Xiuming Zhang
Cancer Control.2023;[Epub] CrossRef - Post-treatment serum triglyceride: An effective biomarker for body fat mass and overall survival in esophageal squamous cell cancer patients treated with chemoradiotherapy
Jiahua Lyu, Ningjing Yang, Wang Guan, Ling Xiao, Xinyu Nie, Long Liang, Hansong Bai, Churong Li, Hao Kuang, Xiao Wang, Tao Li
Frontiers in Nutrition.2022;[Epub] CrossRef - Research Progress of MicroRNA-122 in Obesity and Type 2 Diabetes
路路 潘
Advances in Clinical Medicine.2022; 12(06): 5706. CrossRef - Cancer and Obesity: An Obesity Medicine Association (OMA) Clinical Practice Statement (CPS) 2022
Ethan Lazarus, Harold Edward Bays
Obesity Pillars.2022; 3: 100026. CrossRef - Obesity Fact Sheet in Korea, 2021: Trends in Obesity Prevalence and Obesity-Related Comorbidity Incidence Stratified by Age from 2009 to 2019
Ye Seul Yang, Byoung-Duck Han, Kyungdo Han, Jin-Hyung Jung, Jang Won Son
Journal of Obesity & Metabolic Syndrome.2022; 31(2): 169. CrossRef - Fried food consumption and the risk of pancreatic cancer: A large prospective multicenter study
Guo-Chao Zhong, Qian Zhu, Jian-Ping Gong, Dong Cai, Jie-Jun Hu, Xin Dai, Jun-Hua Gong
Frontiers in Nutrition.2022;[Epub] CrossRef - Intermittent Fasting Is Associated With a Decreased Risk of Age-Related Macular Degeneration
Eun Young Choi, Min Kim, Christopher Seungkyu Lee, Suk Ho Byeon, Sung Soo Kim, Minyoung Lee
American Journal of Ophthalmology.2022; 243: 1. CrossRef - Metformin Mitigated Obesity-Driven Cancer Aggressiveness in Tumor-Bearing Mice
Chun-Jung Chen, Chih-Cheng Wu, Cheng-Yi Chang, Jian-Ri Li, Yen-Chuan Ou, Wen-Ying Chen, Su-Lan Liao, Jiaan-Der Wang
International Journal of Molecular Sciences.2022; 23(16): 9134. CrossRef - Visceral fat area and body fat percentage measured by bioelectrical impedance analysis correlate with glycometabolism
Shuying Li, Shaoping Li, Jie Ding, Weihong Zhou
BMC Endocrine Disorders.2022;[Epub] CrossRef - Insight on the Role of Leptin: A Bridge from Obesity to Breast Cancer
Roberto Buonaiuto, Fabiana Napolitano, Sara Parola, Pietro De Placido, Valeria Forestieri, Giovanna Pecoraro, Alberto Servetto, Luigi Formisano, Pietro Formisano, Mario Giuliano, Grazia Arpino, Sabino De Placido, Carmine De Angelis
Biomolecules.2022; 12(10): 1394. CrossRef - Adipokines as Regulators of Autophagy in Obesity-Linked Cancer
Alin García-Miranda, Alejandra Garcia-Hernandez, Eduardo Castañeda-Saucedo, Napoleon Navarro-Tito, Paola Maycotte
Cells.2022; 11(20): 3230. CrossRef - Roles of hydroxyeicosatetraenoic acids in diabetes (HETEs and diabetes)
Linyue Dong, Heyao Wang, Kaixian Chen, Yiming Li
Biomedicine & Pharmacotherapy.2022; 156: 113981. CrossRef - Changes in the Fecal Metabolome Accompany an Increase in Aberrant Crypt Foci in the Colon of C57BL/6 Mice Fed with a High-Fat Diet
Huawei Zeng, Bryan D. Safratowich, Wen-Hsing Cheng, Andrew D. Magnuson, Matthew J. Picklo
Biomedicines.2022; 10(11): 2891. CrossRef - Targeting Adiponectin in Breast Cancer
Rawan Nehme, Mona Diab-Assaf, Caroline Decombat, Laetitia Delort, Florence Caldefie-Chezet
Biomedicines.2022; 10(11): 2958. CrossRef - Multidisciplinary Progress in Obesity Research
Xiaoqing Lu, Yuxin Jin, Dexin Li, Jingxin Zhang, Jingyan Han, Yin Li
Genes.2022; 13(10): 1772. CrossRef - The association between serum copper and obesity and all-cause mortality: the NHANES 2011–2016
Hongrong Wu, Qingqi Li, Kaifang Zhang, Jianfeng Zhao
Environmental Science and Pollution Research.2022; 30(11): 31395. CrossRef - The crosstalk within the breast tumor microenvironment in type II diabetes: Implications for cancer disparities
Christina S. Ennis, Pablo Llevenes, Yuhan Qiu, Ruben Dries, Gerald V. Denis
Frontiers in Endocrinology.2022;[Epub] CrossRef
 PubReader
PubReader ePub Link
ePub Link Cite
Cite