- Current
- Browse
- Collections
-
For contributors
- For Authors
- Instructions to authors
- Article processing charge
- e-submission
- For Reviewers
- Instructions for reviewers
- How to become a reviewer
- Best reviewers
- For Readers
- Readership
- Subscription
- Permission guidelines
- About
- Editorial policy
Articles
- Page Path
- HOME > Diabetes Metab J > Volume 45(6); 2021 > Article
-
Original ArticleComplications Associations of Plasma Glucagon Levels with Estimated Glomerular Filtration Rate, Albuminuria and Diabetic Kidney Disease in Patients with Type 2 Diabetes Mellitus
-
Hua-Xing Huang1,2
 , Liang-Lan Shen2, Hai-Yan Huang3, Li-Hua Zhao3, Feng Xu3, Dong-Mei Zhang4, Xiu-Lin Zhang5, Tong Chen5, Xue-Qin Wang3, Yan Xie1, Jian-Bin Su3
, Liang-Lan Shen2, Hai-Yan Huang3, Li-Hua Zhao3, Feng Xu3, Dong-Mei Zhang4, Xiu-Lin Zhang5, Tong Chen5, Xue-Qin Wang3, Yan Xie1, Jian-Bin Su3 -
Diabetes & Metabolism Journal 2021;45(6):868-879.
DOI: https://doi.org/10.4093/dmj.2020.0149
Published online: March 23, 2021
1Department of General Medicine, First Affiliated Hospital of Soochow University, Suzhou, China
2Department of Nephrology, Affiliated Hospital 2 of Nantong University, and First People’s Hospital of Nantong City, Nantong, China
3Department of Endocrinology, Affiliated Hospital 2 of Nantong University, and First People’s Hospital of Nantong City, Nantong, China
4Medical Research Center, Affiliated Hospital 2 of Nantong University, and First People’s Hospital of Nantong City, Nantong, China
5Department of Clinical Laboratory, Affiliated Hospital 2 of Nantong University, and First People’s Hospital of Nantong City, Nantong, China
-
Corresponding authors: Yan Xie Department of General Medicine, First Affiliated Hospital of Soochow University, No. 188 Shizi Street, Suzhou 215006, China E-mail: xieyan005@163.com
-
Jian-Bin Su Department of Endocrinology, Affiliated Hospital 2 of Nantong University, and First People’s Hospital of Nantong City, No. 6, Haierxiang North Road, Nantong 226001, China E-mail: sujbzjx@163.com
Copyright © 2021 Korean Diabetes Association
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
-
Background
- Type 2 diabetes mellitus (T2DM) is characterized by elevated fasting glucagon and impaired suppression of postprandial glucagon secretion, which may participate in diabetic complications. Therefore, we investigated the associations of plasma glucagon with estimated glomerular filtration rate (eGFR), albuminuria and diabetic kidney disease (DKD) in T2DM patients.
-
Methods
- Fasting glucagon and postchallenge glucagon (assessed by area under the glucagon curve [AUCgla]) levels were determined during oral glucose tolerance tests. Patients with an eGFR <60 mL/min/1.73 m2 and/or a urinary albumin-to-creatinine ratio (UACR) ≥30 mg/g who presented with diabetic retinopathy were identified as having DKD.
-
Results
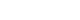
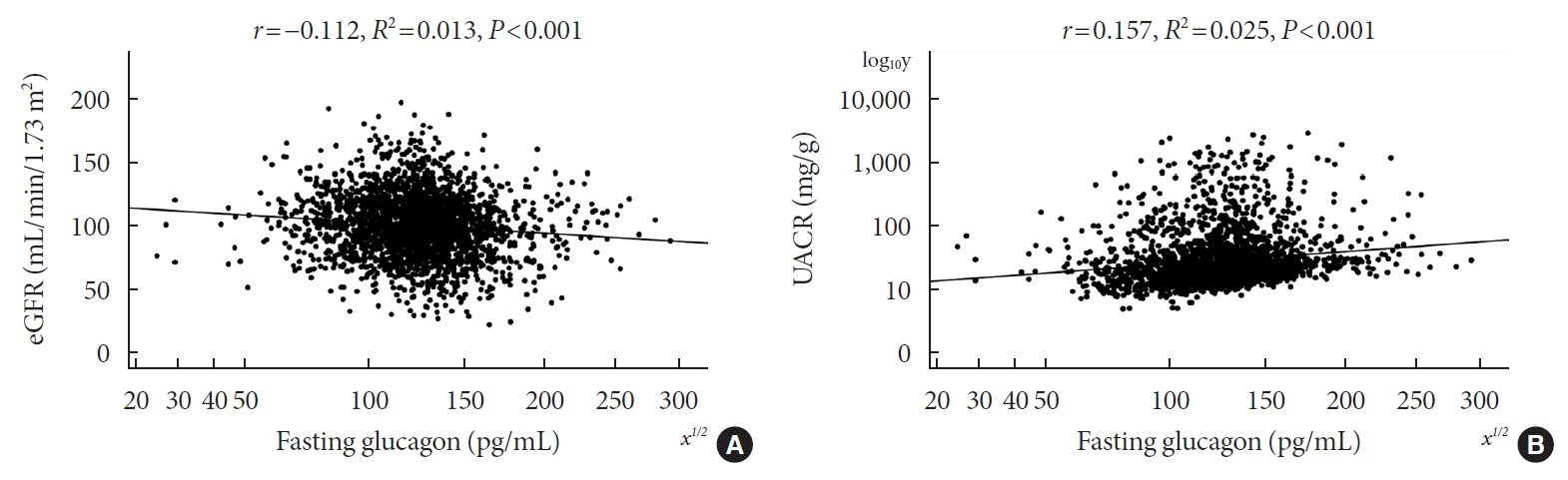
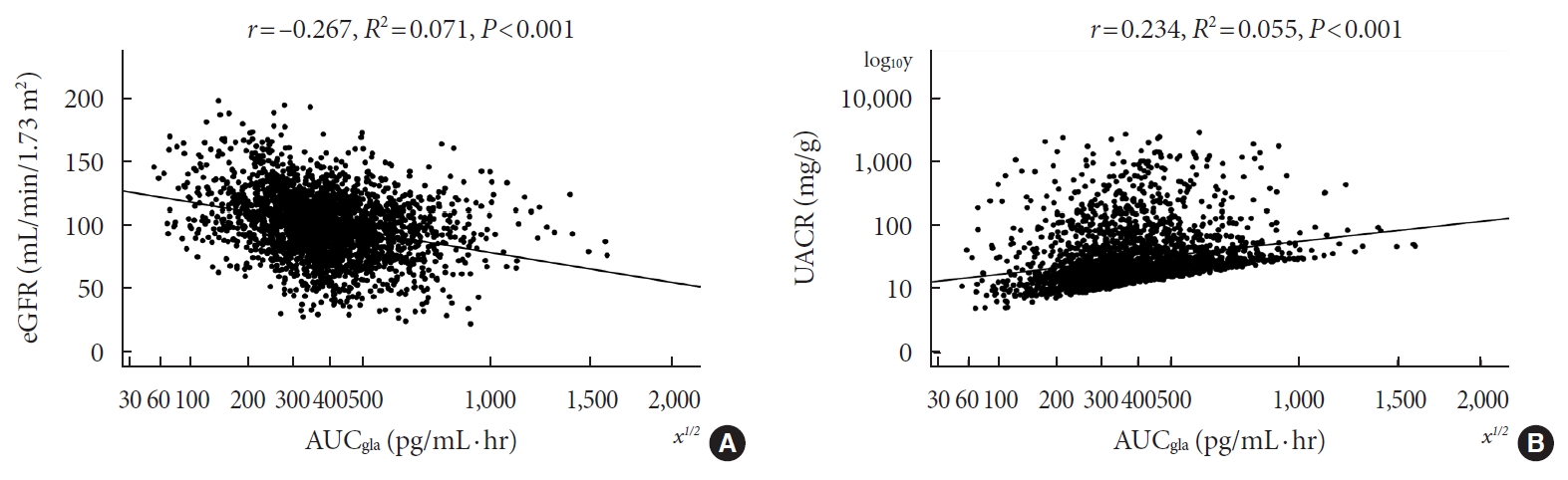
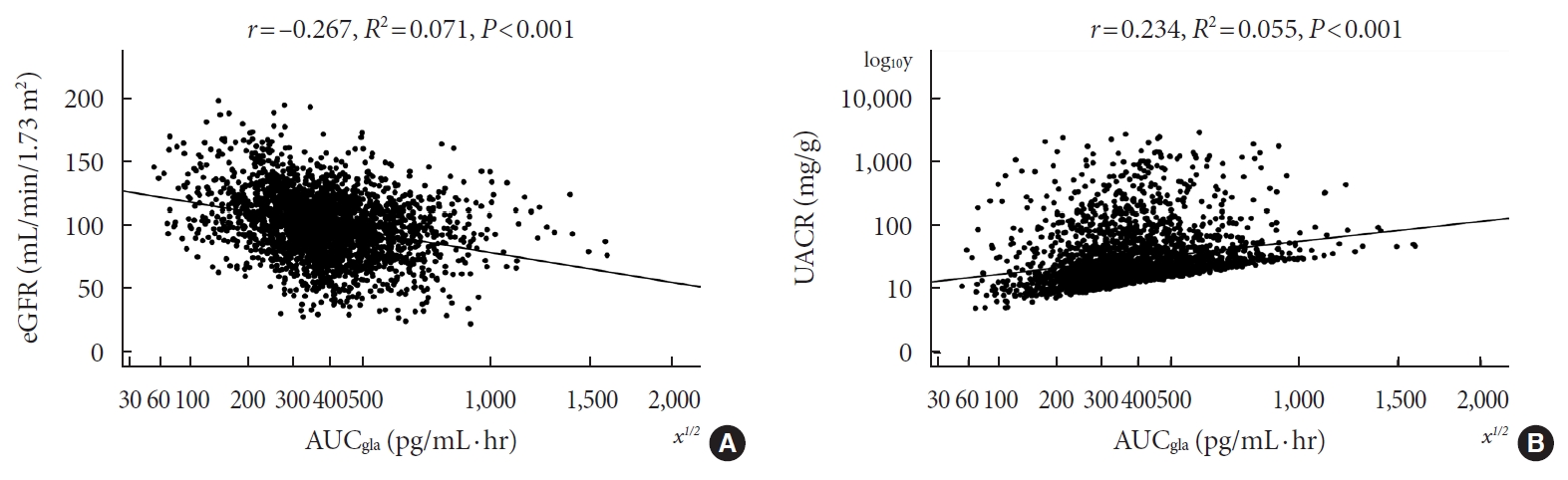
- Of the 2,436 recruited patients, fasting glucagon was correlated with eGFR and UACR (r=–0.112 and r=0.157, respectively; P<0.001), and AUCgla was also correlated with eGFR and UACR (r=–0.267 and r=0.234, respectively; P<0.001). Moreover, 31.7% (n=771) presented with DKD; the prevalence of DKD was 27.3%, 27.6%, 32.5%, and 39.2% in the first (Q1), second (Q2), third (Q3), and fourth quartile (Q4) of fasting glucagon, respectively; and the corresponding prevalence for AUCgla was 25.9%, 22.7%, 33.7%, and 44.4%, respectively. Furthermore, after adjusting for other clinical covariates, the adjusted odds ratios (ORs; 95% confidence intervals) for DKD in Q2, Q3, and Q4 versus Q1 of fasting glucagon were 0.946 (0.697 to 1.284), 1.209 (0.895 to 1.634), and 1.521 (1.129 to 2.049), respectively; the corresponding ORs of AUCgla were 0.825 (0.611 to 1.114), 1.323 (0.989 to 1.769), and 2.066 (1.546 to 2.760), respectively. Additionally, when we restricted our analysis in patients with glycosylated hemoglobin <7.0% (n=471), we found fasting glucagon and AUCgla were still independently associated with DKD.
-
Conclusion
- Both increased fasting and postchallenge glucagon levels were independently associated with DKD in T2DM patients.
- • Hyperglucagonemia may be involved in the diabetic complications, such as diabetic kidney disease (DKD).
- • Increased fasting and postchallenge glucagon levels were mildly correlated with decreased glomerular filtration rate and increased albuminuria.
- • The prevalence of DKD showed an increasing trend with increasing quartiles of either fasting or postchallenge glucagon levels.
- • Hyperglucagonemia in the top quartile of fasting or postchallenge glucagon was associated with an increased risk of DKD.
Highlights
- Diabetic kidney disease (DKD), with a decreased glomerular filtration rate and increased albuminuria as its main clinical characteristics [1], is the most prevalent chronic microvascular disease associated with diabetes [2]. DKD is the major cause of end-stage renal disease (ESRD) worldwide [3], and patients with DKD have a significantly increased risk of cardiovascular and all-cause mortality before they reach ESRD [4]. Moreover, the presence of DKD at baseline can independently predict major adverse cardiovascular events and other microvascular complications in patients with type 2 diabetes mellitus (T2DM) [5]. Thus, ongoing studies worldwide are attempting to investigate how pathological factors and conditions affect DKD to interfere with its incidence and progression in patients with T2DM.
- Patients with T2DM manifest an impaired regulation of glucagon secretion, which leads, importantly, to hyperglycaemia. Specifically, T2DM is characterized by elevated fasting plasma glucagon levels and impaired suppression of glucagon secretion in α-cells following oral source of hyperglycaemia or even paradoxically elevated [6-8]. Abnormally elevated plasma glucagon levels, also termed hyperglucagonemia, may contribute to the progression of diabetes by increasing hepatic glucose production and exacerbating systemic hyperglycaemia, as well as inhibiting the insulin signalling pathway [9,10]. These pathophysiological changes due to hyperglucagonemia may, in turn, take part in the occurrence and progression of diabetesrelated complications, such as DKD. Li et al. [11] demonstrated that long-term exposure to hyperglucagonemia could induce early kidney injury phenotypes of T2DM in mice. Wang et al. [12] showed that T2DM patients with nephropathy exhibited increased plasma glucagon levels. However, there is a lack of comprehensive studies on the associations of plasma glucagon levels with kidney function and DKD with large sample sizes of T2DM patients.
- In the present study, we comprehensively investigated the associations of fasting plasma glucagon levels and postchallenge plasma glucagon levels with estimated glomerular filtration rate (eGFR), albuminuria and the presence of DKD in a large-scale sample of patients with T2DM.
INTRODUCTION
- Patient recruitment
- Patients with T2DM who visited the outpatient department or were admitted as inpatients to the Department of Endocrinology, Affiliated Hospital 2 of Nantong University between 2014 and 2020 were consecutively recruited in this study. The study flowchart is displayed in Supplementary Fig. 1. During the recruitment stage, the major inclusion criteria were as follows: (1) met the T2DM diagnostic criteria released by the American Diabetes Association in 2011 [13]; (2) aged 20 to 75 years; and (3) underwent serum creatinine and cystatin C measurement and urinary albumin-to-creatinine ratio (UACR) evaluation. We also excluded patients with any of the following conditions: (1) type 1 diabetes mellitus, including testing positive for diabetes-associated autoantibodies; (2) history of cancer, especially in the urinary system; (3) chronic viral hepatitis and hepatic cirrhosis; (4) abnormal thyroid function; (5) connective tissue diseases; (6) recent administration of steroid hormones; (7) surgeries associated with the kidneys, urinary bladder and excretory passages; (8) primary glomerular disease such as glomerulonephritis; (9) urinary tract infection, such as pyelonephritis; (10) severe cardiovascular and cerebrovascular diseases, such as myocardial infarction requiring revascularization, heart failure and stroke with sequelae; (11) the use of sodium-glucose cotransporter-2 inhibitors (SGLT-2Is); and (12) ESRD requiring renal replacement therapy. Finally, a total of 2,436 T2DM patients with complete data were pooled for analysis. The performance of our study complied with the principles of the Declaration of Helsinki. Our study received ethics approval from the Medical Ethics Committee at the Affiliated Hospital 2 of Nantong University (2014YX012), with written informed consent received from all patients.
- Data collection
- When the patients visited the Department of Endocrinology, they were interviewed and examined to collect clinical data, including data on medical histories, diabetic duration, age, sex, height, weight, body mass index (BMI), systolic/diastolic blood pressure (SBP/DBP), and prescribed medications. The contemporaneous biochemical and imaging data were extracted from the medical records in the hospital information platform. The medication prescriptions focused on glucose-lowering therapies such as intervention by lifestyle alone, insulin injections, insulin secretagogues, α-glucosidase inhibitors (AGIs), metformin, thiazolidinedione (i.e., pioglitazone), glucagon-like peptide-1 receptor agonists (GLP-1RAs), dipeptidyl peptidase-4 inhibitors (DPP-4Is), and SGLT-2Is, and other recent medications such as antihypertensive drugs, anticoagulants, statins, and steroid hormones. Past medical histories included other self-reported types of diabetes, hypertension history, chronic hepatitis, and hepatic cirrhosis (i.e., hepatitis B or C virus infection), inflammation in the urinary system (i.e., pyelonephritis and cystitis), surgeries in the urinary system (kidneys, urinary bladder, and excretory passages), a history of cancer (especially in the urinary system), severe cardiovascular and cerebrovascular diseases (i.e., myocardial infarction requiring revascularization, heart failure of New York Heart Association functional class III or IV, and stroke with sequelae), thyroid dysfunction (i.e., hyperthyroidism and hypothyroidism), etc.
- DKD assessment
- Fasting venous blood samples were also drawn for the measurement of serum creatinine and cystatin C, and eGFR was calculated by the combined creatinine-cystatin C equation from Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) (2012) [14]. Urinary albumin and creatinine were determined to calculate the UACR. Patients with an eGFR <60 mL/min/1.73 m2 and/or a UACR ≥30 mg/g [15] who presented with diabetic retinopathy [16] were identified as having DKD. Diabetic retinopathy was identified by one of the following: (1) a history of diabetic retinopathy; (2) previously received photocoagulation or vitrectomy therapy for diabetic retinopathy; and (3) received nonmydriatic digital fundus photographs to screen for diabetic retinopathy (TOPCON, TRC-NW400). The photographs were analysed by experienced ophthalmologists to identify diabetic retinopathy, and fluorescein angiography was performed on the equivocal image.
- Evaluation of plasma glucagon levels and other biomarkers during the oral glucose tolerance test
- All patients were guided to consume their usual stable diet for three days before the oral glucose tolerance test (OGTT) and to suspend all glucose-lowering therapies one day before the OGTT. In the early morning on the test day, venous blood samples were obtained at fasting (0 minute) and at 30, 60, 120, and 180 minutes after 75-g glucose loading for the synchronous detection of glucose, C-peptide, and glucagon levels. Insulin sensitivity was derived from the homeostasis model assessment of insulin resistance using C-peptide (ISHOMAcp)=22.5/(fasting glucose×fasting C-peptide) [17]. Endogenous β-cell function was assessed by the area under the C-peptide curve (AUCcp) [18,19]. Postchallenge glucose levels were assessed by the area under the glucose curve (AUCglu). Fasting plasma glucagon levels were determined, and postchallenge plasma glucagon levels were assessed by the area under the glucagon curve (AUCgla) during the OGTT.
- Laboratory tests
- The level of urinary albumin was detected with the immunoturbidimetry method (Immage 800; Beckman Coulter, Brea, CA, USA), and urinary creatinine was detected with the enzymatic method (Model 7600; Hitachi, Tokyo, Japan). Regarding the biochemical parameters during the OGTT, glucose levels were detected with the oxidase method (Model 7600; Hitachi), serum C-peptide levels were detected with the chemiluminescence method (DxI 800; Beckman Coulter), and plasma glucagon levels were detected with the radioimmunoassay method in an automated γ-counter (GC-1200; USTC Zonkia, Hefei, China). On the test day, fasting venous blood samples were also obtained from all patients for the detection of clinical biochemical indices. Serum creatinine (using the enzymatic method), cystatin C (using the latex-enhanced immunoturbidimetric assay method), uric acid (UA), triglyceride (TG), total cholesterol (TC), low-density lipoprotein cholesterol (LDL-C) and high-density lipoprotein cholesterol (HDL-C) were detected with an automated biochemical analyser (Model 7600; Hitachi). The glycosylated hemoglobin (HbA1c) levels were detected with an ion exchange-based HPLC method (D-10; Bio-Rad, Hercules, CA, USA).
- Statistical analyses
- Clinical variables are displayed for all patients with T2DM and for two subgroups, those with and without DKD, and are expressed as mean±standard deviation for normally distributed data, as the medians (interquartile ranges) for skewed continuous data and as the frequencies (percentages) for categorical data. If a variable was skewed, it would receive a logarithm transformation for further statistical analysis.
- We used Student’s t-tests and the chi-squared test to detect differences in continuous data and categorical data, respectively, between the two subgroups with and without DKD. Corresponding test statistics, including F values and x2 values, were also provided.
- Bivariate correlations of plasma glucagon levels with eGFR and UACR were analysed by means of Pearson’s correlation tests, and scatter plots for these correlations are graphically displayed. Moreover, to show the degree of influence of plasma glucagon levels on eGFR and UACR, we used multivariable linear regression analyses to detect the mean differences (B; 95% confidence interval [CI]) in eGFR or UACR among the patients in different plasma glucagon level quartiles, with the first quartile (Q1) set as the reference value.
- Furthermore, to investigate the impact of fasting or postchallenge glucagon levels on the risk of DKD in patients with T2DM, we used two multivariate logistic regression analyses to explore the odds ratios (ORs [95% CIs]) for DKD in different levels of fasting or postchallenge glucagon. The first regression model explored the associations of the second (Q2), third (Q3), and fourth quartiles (Q4) of fasting plasma glucagon levels with DKD relative to that of the first quartile (Q1). The second regression model explored the associations of Q2, Q3, and Q4 of postchallenge glucagon levels (AUCgla) with DKD relative to Q1. The potential confounding factors were adjusted in the models. SPSS for Windows standard version 19.0 (IBM Co., Armonk, NY, USA) was used to input and analyse the data, and statistical significance was identified when the P value was less than 0.05.
METHODS
- Table 1 summarizes the clinical characteristics of the recruited patients for all patients and for the two subgroups with and without DKD. Of the recruited patients with T2DM, 31.7% (n=771) presented with DKD, and patients with DKD were characterized by increased fasting plasma glucagon, AUCgla and AUCglu and decreased ISHOMAcp when compared to patients without DKD, but AUCcp values were comparable between the two subgroups. Clinical variables such as age, the proportion of females, BMI, SBP, DBP, diabetic duration, the prevalence of hypertension, statin use, TG, TC, UA, HbA1c, and postchallenge 2-hour glucose were higher in patients with DKD than in patients without DKD, while HDL-C, serum albumin and haemoglobin were lower in patients with DKD than in patients without DKD. LDL-C did not exhibit any difference between the subgroups with and without DKD. Comparisons of the use of glucose-lowering therapies showed that patients with DKD tended to have a higher frequency of insulin treatments and a lower frequency of metformin use than patients without DKD, whereas the use of lifestyle intervention, insulin secretagogues, pioglitazone, AGIs, DPP-4Is, and GLP-1RAs was comparable between the two subgroups.
- Pearson’s correlation tests showed that fasting plasma glucagon was negatively correlated with eGFR (r=–0.112, P<0.001) and positively correlated with UACR (r=0.157, P<0.001), and AUCgla was also negatively correlated with eGFR (r=–0.267, P<0.001) and positively correlated with UACR (r=0.234, P<0.001). Scatter plots for these correlations are graphically displayed in Figs 1 and 2. Moreover, we used multivariable linear regression analyses to show the degree of the influence of plasma glucagon levels on eGFR and UACR in all T2DM patients. After adjusting for other clinical risk factors, the adjusted mean changes (B) in eGFR and log10UACR of the patients in Q4 versus Q1 of fasting plasma glucagon were –5.087 mL/min/1.73 m2 (95% CI, –7.383 to –2.790) and 0.124 mg/g (95% CI, 0.076 to 0.171), respectively (Table 2). The corresponding mean changes in eGFR and log10UACR of the patients in Q4 versus Q1 of AUCgla were –13.89 mL/min/1.73 m2 (95% CI, –16.18 to –11.60) and 0.188 mg/g (95% CI, 0.140 to 0.236), respectively (Table 3).
- Of the recruited patients with T2DM, 31.7% (n=771) presented with DKD, and the prevalence of DKD increased from 27.3%, 27.6%, and 32.5% to 39.2% from Q1, Q2, and Q3 to Q4 for fasting plasma glucagon (P for trend <0.001) (Table 4). Compared with the patients in Q1 of fasting plasma glucagon, those in Q2, Q3, and Q4 of fasting plasma glucagon experienced ORs of DKD of 1.016 (95% CI, 0.789 to 1.308), 1.283 (95% CI, 1.003 to 1.640), and 1.722 (95% CI, 1.353 to 2.191), respectively (P for trend <0.001) (Table 4). Moreover, after adjusting for glycaemic status and other clinical risk factors via multivariate logistic regression analysis, the corresponding ORs for DKD of patients in Q2, Q3, and Q4 versus those in Q1 of fasting plasma glucagon were 0.946 (95% CI, 0.697 to 1.284), 1.209 (95% CI, 0.895 to 1.634), and 1.521 (95% CI, 1.129 to 2.049), respectively (P for trend <0.001) (Table 2).
- Furthermore, of the recruited patients with T2DM, the prevalence of DKD increased from 25.9%, 22.7%, and 33.7% to 44.4% from Q1, Q2, and Q3 to Q4 of AUCgla (P for trend <0.001) (Table 4). Compared with the patients in Q1 of AUCgla, those in Q2, Q3, and Q4 of AUCgla experienced ORs of DKD of 0.841 (95% CI, 0.647 to 1.093), 1.454 (95% CI, 1.134 to 1.863), and 2.283 (95% CI, 1.791 to 2.910), respectively (P for trend <0.001) (Table 4). Moreover, after adjusting for glycaemic status and other clinical risk factors via multivariate logistic regression analysis, the corresponding ORs for DKD of patients in Q2, Q3, and Q4 versus those in Q1 of AUCgla were 0.825 (95% CI, 0.611 to 1.114), 1.323 (95% CI, 0.989 to 1.769), and 2.066 (95% CI, 1.546 to 2.760), respectively (P for trend <0.001) (Table 4).
- Additionally, when we restricted our analysis to the recruited patients with HbA1c <7.0% (n=471), we found that fasting glucagon and AUCgla levels were still independently associated with kidney damage indices (eGFR and UACR), and increased fasting glucagon and AUCgla levels were still risk factors for DKD in these patients with well-controlled T2DM (Supplementary Tables 1-4, Supplementary Figs 2 and 3).
RESULTS
- In the present study, we explored the associations of fasting and postchallenge plasma glucagon levels with the presence of DKD in a large sample of patients with T2DM (n=2,436). The strengths of our study are as follows: first, both increased fasting plasma glucagon levels and postchallenge plasma glucagon levels are revealed to be mildly correlated with decreased eGFR and increased UACR; second, 31.7% (n=771) of the recruited patients presented with DKD, and the prevalence of DKD showed an increasing trend with increasing quartiles of either fasting plasma glucagon levels or postchallenge plasma glucagon levels; third, patients from Q4 of fasting plasma glucagon levels exhibited a 1.521-fold (95% CI, 1.129 to 2.049) increased DKD risk compared with those in Q1 after adjustment for other confounders, and the corresponding adjusted DKD risk for patients from Q4 of postchallenge plasma glucagon levels was 2.066-fold (95% CI, 1.546 to 2.760); and fourth, when we restricted our analysis to the recruited patients with HbA1c <7.0% (n=471), we found fasting and postchallenge plasma glucagon levels were still independently associated with DKD. Collectively, hyperglucagonemia in the top quartile of fasting or postchallenge glucagon levels is associated with an increased risk of DKD in patients with T2DM independent of glucose levels.
- Ongoing work worldwide is seeking markers of and risk factors for DKD. The incidence and progression of DKD is in the background of diabetes; therefore, glycaemic disorders may be the initiator and driving force for DKD, and other various cardiovascular risks may be the promoters for DKD [20]. The irreversible risk factors included older age, sex, ethnicity, a family history of DKD and a long duration of diabetes, and the reversible factors included smoking, hypertension, dyslipidaemia, hypercoagulation, insulin resistance, albuminuria, anaemia, etc. [21-23]. These risk factors subsequently may account for the immune-inflammatory responses in the progression of DKD. In addition, in the immune-inflammatory response pathways, innate immune cells (mast cells, etc.), proinflammatory cytokines (interleukin-1, -6, -18, and tumour necrosis factor-α, etc.), chemokines (monocyte chemoattractant protein-1, etc.), cell adhesion molecules, and growth factor (transforming growth factor-β, etc.) have been implicated in the pathogenesis of DKD [24-26]. These responses ultimately may lead to irreversible changes in the function and structure of DKD, such as interstitial fibrosis and glomerulosclerosis in the kidney [27]. Moreover, impaired regulation of glucagon secretion and islet β-cell dysfunction are associated with DKD. Deficiency in endogenous β-cell function assessed by AUCcp is linked to an increased prevalence of nephropathy in the population with T2DM [18], and T2DM patients with nephropathy exhibited abnormally elevated glucagon levels [12]. In our present study, hyperglucagonemia at both fasting status and postchallenge status was found to be associated with an increased prevalence of DKD in patients with T2DM, independent of glycaemic controls and other clinical risk factors.
- Hyperglucagonemia is at least partially responsible for disorders of glucose, lipid and amino acid metabolism, which, in turn, take part in the pathophysiology of diabetes, obesity and nonalcoholic fatty liver disease [28] and ultimately lead to various adverse health outcomes. Relative hyperglucagonemia, under the condition of deficiency in insulin secretion, is involved in the pathogenesis of glycaemic variability measured by continuous glucose monitoring and serum hyperglycaemia in diabetes [29-31]. These glycaemic disorders are closely associated with the morbidity and mortality of cardiovascular diseases [32,33]. Hyperglucagonemia could also contribute to adverse cardiovascular outcomes through lipid metabolism pathways. Ali et al. [34] demonstrated that glucagon administration could increase long-chain acylcarnitines by triggering an increase in β-oxidation in ischaemic mouse hearts, which, in turn, promoted cardiac ischaemic injury in mice and led to cardiomyocyte apoptosis. In contrast, Gao et al. [35] showed that the blockade of the glucagon receptor (GCGR) can ameliorate cardiac hypertrophy and fibrotic remodelling and attenuate exacerbation in heart failure after myocardial infarction in mice. In addition, the blockade of GCGR-mediated improvement in AMP-activated protein kinase (AMPK)-induced lipid oxidation and protection against cardiac dysfunction is independent of the correction of glycaemia [36]. Moreover, elevated levels of glucagon were correlated with serum levels of total amino acids and several non-branched-chain amino acids, such as glutamine and asparagine [37]. These abnormal changes in glutamine and asparagine are likely metabolic markers for chronic kidney disease [38,39]. Additionally, acute elevation of plasma glucagon has been documented to directly induce glomerular hyperfiltration—a hallmark of early glomerular injury in T2DM [40], and long-term exposure to hyperglucagonemia could also induce kidney injury phenotypes of T2DM [11]. In population-based studies, T2DM patients with nephropathy exhibited increased glucagon levels at different times after glucose loading [12], and fasting glucagon levels were elevated linearly with the progression of kidney dysfunction in patients with T2DM [41]. In our present study of 2,436 patients with T2DM, we used diabetic retinopathy to aid in the diagnosis of DKD on the basis of decreased eGFR and/or increased UACR, which may avoid the limitations of eGFR and albuminuria. We found that increased fasting and postchallenge glucagon levels were not only associated with the kidney dysfunction index—a decline in eGFR—but also associated with damage to the glomerular filtration barrier—increased albuminuria. Patients from the upper quartile of fasting glucagon levels or postchallenge glucagon levels experienced an increased risk for DKD compared to those from the lower quartile. Hyperglucagonemia in the upper quartile of fasting or postchallenge glucagon levels accounts for an increased risk of DKD in patients with T2DM.
- Some underlying mechanisms have been suggested in the relationship between hyperglucagonemia and DKD. In the context of diabetes, disorder in glycaemic metabolism is a critical and initiating risk factor for DKD. Elevated fasting glucagon levels contribute to fasting hyperglycaemia by increasing glycogenolysis and gluconeogenesis in the liver, and impaired suppression of glucagon secretion may contribute to postprandial hyperglycaemia [30]. The subsequent fluctuations between fasting and postprandial hyperglycaemia may contribute to glycaemic variability throughout the day. All these features in glycaemic disorders can lead to the incidence and progression of DKD [42,43]. Moreover, hyperglucagonemia can also cause kidney damage in a glucose-independent manner. Increased plasma glucagon levels, via direct overactivation of GCGR-mediated signalling pathways, induce the phosphorylation of extracellular signal-regulated kinase 1/2 (Erk1/2), finally stimulating mesangial cell (MC) growth and hypertrophy and resulting in glomerular mesangial expansion and extracellular matrix deposition [44]. Glucagon can induce the phosphorylation of ERK1/2 in MCs by three signalling pathways, including the cAMP-dependent protein kinase A (PKA) pathway, the phospholipase C (PLC)/inositol 1,4,5-trisphosphate (IP3)/Ca2+ pathways [45], and the pathway that stimulates angiotensin II (Ang II) formation and interacts with the Ang II receptor [46]. Furthermore, GCGR antagonists are expected to block the metabolic and growth effects of hyperglucagonemia in T2DM [44,47].
- Several limitations of the study must be addressed. First, this study is based on the analysis of cross-sectional data, which may not necessarily indicate the existence of a causal link between increased glucagon levels and DKD. A follow-up study is needed to compensate for this limitation. Second, our present study is influenced by the heterogeneities of T2DM, in that the levels of insulin and glucagon may be influenced by glucose-lowering therapies. We adjusted glucose-lowering therapies during the analyses and tried to compensate for the limitation as much as possible. Third, the gold standard of the diagnosis of DKD is based on renal biopsy, but it is too labour-intensive and expensive to be applied in large clinical studies. Fourth, we assessed only glucagon secretion of α-cells in response to oral glucose, and postprandial glucagon levels may be different when responding to the different nutrients.
- In conclusion, both increased fasting plasma glucagon levels and postchallenge plasma glucagon levels following an OGTT were independently associated with DKD in patients with T2DM.
DISCUSSION
Supplementary Materials
Supplementary Table 1.
Supplementary Table 2.
Supplementary Table 3.
Supplementary Table 4.
Supplementary Fig. 1.
Supplementary Fig. 2.
Supplementary Fig. 3.
-
CONFLICTS OF INTEREST
No potential conflict of interest relevant to this article was reported.
-
AUTHOR CONTRIBUTIONS
Conception or design: H.X.H., Y.X., J.B.S.
Acquisition, analysis, or interpretation of data: L.L.S., H.Y.H., L.H.Z., F.X., D.M.Z., X.L.Z., T.C., X.Q.W.
Drafting the work or revising: H.X.H., Y.X., J.B.S.
Final approval of the manuscript: H.X.H., L.L.S., H.Y.H., L.H.Z., F.X., D.M.Z., X.L.Z., T.C., X.Q.W., Y.X., J.B.S.
-
FUNDING
The study was funded by the Social Development Projects of Nantong (HS2012028, MS22015065, MS12019019), the Medical Research Project of Nantong Health Commission (MB201-9010, 2020JCC010), and the Medical Research Project of Jiangsu Health Commission (QNRC2016408).
NOTES
-
Acknowledgements
- None
Values are presented as mean±standard deviation, number (%), or median (interquartile range). Student’s t-test and chi-square test were applied to detect differences in their corresponding type of data, and corresponding test statistics (F and x2 values) were also provided.
DKD, diabetic kidney disease; BMI, body mass index; SBP, systolic blood pressure; DBP, diastolic blood pressure; AGI, α-glucosidase inhibitor; DPP-4I, dipeptidyl peptidase-4 inhibitor; GLP-1RA, glucagon-like peptide-1 receptor agonist; TG, triglyceride; TC, total cholesterol; HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol; UA, uric acid; ISHOMAcp, homeostasis model assessment of insulin resistance using C-peptide; AUCcp, area under the C-peptide curve; AUCglu, area under the glucose curve; HbA1c, glycosylated hemoglobin; AUCgla, area under the glucagon curve; eGFR, estimated glomerular filtration rate; UACR, urinary albumin-to-creatinine ratio.
Values are presented as range, number, mean±standard deviation, or mean difference (95% confidence interval).
Model 0: crude; Model 1: adjusted for age, sex, diabetic duration, body mass index, systolic blood pressure, diastolic blood pressure, statins medication, and hypertension; Model 2: additionally adjusted for lipid profiles, serum uric acid, serum albumin, hemoglobin, homeostasis model assessment of insulin resistance using C-peptide, area under the C-peptide curve, area under the glucose curve, postchallenge 2-hour glucose, and glycosylated hemoglobin; Model 3: additionally adjusted for glucose-lowering therapies.
CI, confidence interval; T2DM, type 2 diabetes mellitus; eGFR, estimated glomerular filtration rate; UACR, urinary albumin-to-creatinine ratio.
Values are presented as range, number, mean±standard deviation, or mean difference (95% confidence interval).
Model 0: crude; Model 1: adjusted for age, sex, diabetic duration, body mass index, systolic blood pressure, diastolic blood pressure, statins medication, and hypertension; Model 2: additionally adjusted for lipid profiles, serum uric acid, serum albumin, hemoglobin, homeostasis model assessment of insulin resistance using C-peptide, area under the C-peptide curve, area under the glucose curve, postchallenge 2-hour glucose, and glycosylated hemoglobin; Model 3: additionally adjusted for glucose-lowering therapies.
CI, confidence interval; AUCgla, area under the glucagon curve; T2DM, type 2 diabetes mellitus; eGFR, estimated glomerular filtration rate; UACR, urinary albumin-to-creatinine ratio.
Values are presented as range, number (%), or odds ratio (95% confidence interval).
Model 0: crude; Model 1: adjusted for age, sex, diabetic duration, body mass index, systolic blood pressure, diastolic blood pressure, statins medication, and hypertension; Model 2: additionally adjusted for lipid profiles, serum uric acid, serum albumin, hemoglobin, homeostasis model assessment of insulin resistance using C-peptide, area under the C-peptide curve, area under the glucose curve, postchallenge 2-hour glucose, and glycosylated hemoglobin; Model 3: additionally adjusted for glucose-lowering therapies.
OR, odds ratio; CI, confidence interval; DKD, diabetic kidney disease; T2DM, type 2 diabetes mellitus; AUCgla, area under the glucagon curve.
- 1. Afkarian M, Zelnick LR, Hall YN, Heagerty PJ, Tuttle K, Weiss NS, et al. Clinical manifestations of kidney disease among US adults with diabetes, 1988-2014. JAMA 2016;316:602-10.ArticlePubMedPMC
- 2. Koye DN, Magliano DJ, Nelson RG, Pavkov ME. The global epidemiology of diabetes and kidney disease. Adv Chronic Kidney Dis 2018;25:121-32.ArticlePubMed
- 3. Harding JL, Pavkov ME, Magliano DJ, Shaw JE, Gregg EW. Global trends in diabetes complications: a review of current evidence. Diabetologia 2019;62:3-16.ArticlePubMedPDF
- 4. Fox CS, Matsushita K, Woodward M, Bilo HJ, Chalmers J, Heerspink HJ, et al. Associations of kidney disease measures with mortality and end-stage renal disease in individuals with and without diabetes: a meta-analysis. Lancet 2012;380:1662-73.ArticlePubMedPMC
- 5. Mohammedi K, Woodward M, Marre M, Colagiuri S, Cooper M, Harrap S, et al. Comparative effects of microvascular and macrovascular disease on the risk of major outcomes in patients with type 2 diabetes. Cardiovasc Diabetol 2017;16:95.ArticlePubMedPMCPDF
- 6. Wewer Albrechtsen NJ, Faerch K, Jensen TM, Witte DR, Pedersen J, Mahendran Y, et al. Evidence of a liver-alpha cell axis in humans: hepatic insulin resistance attenuates relationship between fasting plasma glucagon and glucagonotropic amino acids. Diabetologia 2018;61:671-80.ArticlePubMedPDF
- 7. Knop FK, Vilsboll T, Madsbad S, Holst JJ, Krarup T. Inappropriate suppression of glucagon during OGTT but not during isoglycaemic i.v. glucose infusion contributes to the reduced incretin effect in type 2 diabetes mellitus. Diabetologia 2007;50:797-805.ArticlePubMedPDF
- 8. Lund A, Bagger JI, Christensen M, Grondahl M, van Hall G, Holst JJ, et al. Higher endogenous glucose production during OGTT vs isoglycemic intravenous glucose infusion. J Clin Endocrinol Metab 2016;101:4377-84.ArticlePubMedPDF
- 9. Wewer Albrechtsen NJ, Pedersen J, Galsgaard KD, Winther-Sorensen M, Suppli MP, Janah L, et al. The liver-α-cell axis and type 2 diabetes. Endocr Rev 2019;40:1353-66.ArticlePubMedPDF
- 10. Holst JJ, Holland W, Gromada J, Lee Y, Unger RH, Yan H, et al. Insulin and glucagon: partners for life. Endocrinology 2017;158:696-701.ArticlePubMedPMC
- 11. Li XC, Liao TD, Zhuo JL. Long-term hyperglucagonaemia induces early metabolic and renal phenotypes of type 2 diabetes in mice. Clin Sci (Lond) 2008;114:591-601.ArticlePubMedPMCPDF
- 12. Wang X, Yang J, Chang B, Shan C, Xu Y, Zheng M, et al. Glucagon secretion is increased in patients with type 2 diabetic nephropathy. J Diabetes Complications 2016;30:488-93.ArticlePubMed
- 13. American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care 2011;34(Suppl 1):S62-9.ArticlePubMedPMCPDF
- 14. Inker LA, Schmid CH, Tighiouart H, Eckfeldt JH, Feldman HI, Greene T, et al. Estimating glomerular filtration rate from serum creatinine and cystatin C. N Engl J Med 2012;367:20-9.ArticlePubMedPMC
- 15. De Boer IH, Rue TC, Hall YN, Heagerty PJ, Weiss NS, Himmelfarb J. Temporal trends in the prevalence of diabetic kidney disease in the United States. JAMA 2011;305:2532-9.ArticlePubMedPMC
- 16. Gosmanov AR, Wall BM, Gosmanova EO. Diagnosis and treatment of diabetic kidney disease. Am J Med Sci 2014;347:406-13.ArticlePubMed
- 17. Radaelli T, Farrell KA, Huston-Presley L, Amini SB, Kirwan JP, McIntyre HD, et al. Estimates of insulin sensitivity using glucose and C-Peptide from the hyperglycemia and adverse pregnancy outcome glucose tolerance test. Diabetes Care 2010;33:490-4.ArticlePubMedPDF
- 18. Zhao L, Ma J, Wang S, Xie Y. Relationship between β-cell function, metabolic control, and microvascular complications in type 2 diabetes mellitus. Diabetes Technol Ther 2015;17:29-34.ArticlePubMed
- 19. Su JB, Wu YY, Xu F, Wang X, Cai HL, Zhao LH, et al. Serum complement C3 and islet β-cell function in patients with type 2 diabetes: a 4.6-year prospective follow-up study. Endocrine 2020;67:321-30.ArticlePubMedPDF
- 20. Macisaac RJ, Ekinci EI, Jerums G. Markers of and risk factors for the development and progression of diabetic kidney disease. Am J Kidney Dis 2014;63(2 Suppl 2):S39-62.ArticlePubMed
- 21. Guo K, Zhang L, Zhao F, Lu J, Pan P, Yu H, et al. Prevalence of chronic kidney disease and associated factors in Chinese individuals with type 2 diabetes: cross-sectional study. J Diabetes Complications 2016;30:803-10.ArticlePubMed
- 22. Alicic RZ, Rooney MT, Tuttle KR. Diabetic kidney disease: challenges, progress, and possibilities. Clin J Am Soc Nephrol 2017;12:2032-45.PubMedPMC
- 23. Alicic RZ, Johnson EJ, Tuttle KR. Inflammatory mechanisms as new biomarkers and therapeutic targets for diabetic kidney disease. Adv Chronic Kidney Dis 2018;25:181-91.ArticlePubMed
- 24. Pichler R, Afkarian M, Dieter BP, Tuttle KR. Immunity and inflammation in diabetic kidney disease: translating mechanisms to biomarkers and treatment targets. Am J Physiol Renal Physiol 2017;312:F716-31.ArticlePubMed
- 25. Looker HC, Mauer M, Nelson RG. Role of kidney biopsies for biomarker discovery in diabetic kidney disease. Adv Chronic Kidney Dis 2018;25:192-201.ArticlePubMedPMC
- 26. Sharma D, Bhattacharya P, Kalia K, Tiwari V. Diabetic nephropathy: new insights into established therapeutic paradigms and novel molecular targets. Diabetes Res Clin Pract 2017;128:91-108.ArticlePubMed
- 27. Umanath K, Lewis JB. Update on diabetic nephropathy: core curriculum 2018. Am J Kidney Dis 2018;71:884-95.ArticlePubMed
- 28. Janah L, Kjeldsen S, Galsgaard KD, Winther-Sorensen M, Stojanovska E, Pedersen J, et al. Glucagon receptor signaling and glucagon resistance. Int J Mol Sci 2019;20:3314.ArticlePubMedPMC
- 29. Takahashi N, Chujo D, Kajio H, Ueki K. Contribution of pancreatic α-cell function to insulin sensitivity and glycemic variability in patients with type 1 diabetes. J Diabetes Investig 2019;10:690-8.ArticlePubMedPDF
- 30. Dunning BE, Gerich JE. The role of alpha-cell dysregulation in fasting and postprandial hyperglycemia in type 2 diabetes and therapeutic implications. Endocr Rev 2007;28:253-83.PubMed
- 31. Cryer PE. Minireview: glucagon in the pathogenesis of hypoglycemia and hyperglycemia in diabetes. Endocrinology 2012;153:1039-48.ArticlePubMedPDF
- 32. Kilpatrick ES, Rigby AS, Atkin SL. Mean blood glucose compared with HbA1c in the prediction of cardiovascular disease in patients with type 1 diabetes. Diabetologia 2008;51:365-71.ArticlePubMedPDF
- 33. Takao T, Matsuyama Y, Yanagisawa H, Kikuchi M, Kawazu S. Association between HbA1c variability and mortality in patients with type 2 diabetes. J Diabetes Complications 2014;28:494-9.ArticlePubMed
- 34. Ali S, Ussher JR, Baggio LL, Kabir MG, Charron MJ, Ilkayeva O, et al. Cardiomyocyte glucagon receptor signaling modulates outcomes in mice with experimental myocardial infarction. Mol Metab 2014;4:132-43.ArticlePubMedPMC
- 35. Gao C, Ren SV, Yu J, Baal U, Thai D, Lu J, et al. Glucagon receptor antagonism ameliorates progression of heart failure. JACC Basic Transl Sci 2019;4:161-72.ArticlePubMedPMC
- 36. Sharma AX, Quittner-Strom EB, Lee Y, Johnson JA, Martin SA, Yu X, et al. Glucagon receptor antagonism improves glucose metabolism and cardiac function by promoting AMP-mediated protein kinase in diabetic mice. Cell Rep 2018;22:1760-73.ArticlePubMedPMC
- 37. Wewer Albrechtsen NJ, Junker AE, Christensen M, Haedersdal S, Wibrand F, Lund AM, et al. Hyperglucagonemia correlates with plasma levels of non-branched-chain amino acids in patients with liver disease independent of type 2 diabetes. Am J Physiol Gastrointest Liver Physiol 2018;314:G91-6.ArticlePubMed
- 38. Kim JA, Choi HJ, Kwon YK, Ryu DH, Kwon TH, Hwang GS. 1H NMR-based metabolite profiling of plasma in a rat model of chronic kidney disease. PLoS One 2014;9:e85445.ArticlePubMedPMC
- 39. Hanifa MA, Skott M, Maltesen RG, Rasmussen BS, Nielsen S, Frokiaer J, et al. Tissue, urine and blood metabolite signatures of chronic kidney disease in the 5/6 nephrectomy rat model. Metabolomics 2019;15:112.ArticlePubMedPDF
- 40. Ahloulay M, Dechaux M, Laborde K, Bankir L. Influence of glucagon on GFR and on urea and electrolyte excretion: direct and indirect effects. Am J Physiol 1995;269(2 Pt 2):F225-35.ArticlePubMed
- 41. Liu JJ, Liu S, Gurung RL, Chan C, Ang K, Tang WE, et al. Relationship between fasting plasma glucagon level and renal function: a cross-sectional study in individuals with type 2 diabetes. J Endocr Soc 2018;3:273-83.ArticlePubMedPMCPDF
- 42. Ying C, Zhou X, Chang Z, Ling H, Cheng X, Li W. Blood glucose fluctuation accelerates renal injury involved to inhibit the AKT signaling pathway in diabetic rats. Endocrine 2016;53:81-96.ArticlePubMedPDF
- 43. Wang C, Song J, Ma Z, Yang W, Li C, Zhang X, et al. Fluctuation between fasting and 2-H postload glucose state is associated with chronic kidney disease in previously diagnosed type 2 diabetes patients with HbA1c ≥ 7%. PLoS One 2014;9:e102941.ArticlePubMedPMC
- 44. Li XC, Zhuo JL. Targeting glucagon receptor signalling in treating metabolic syndrome and renal injury in type 2 diabetes: theory versus promise. Clin Sci (Lond) 2007;113:183-93.ArticlePubMedPMCPDF
- 45. Li XC, Carretero OA, Shao Y, Zhuo JL. Glucagon receptor-mediated extracellular signal-regulated kinase 1/2 phosphorylation in rat mesangial cells: role of protein kinase A and phospholipase C. Hypertension 2006;47:580-5.ArticlePubMed
- 46. Li XC, Carretero OA, Zhuo JL. Cross-talk between angiotensin II and glucagon receptor signaling mediates phosphorylation of mitogen-activated protein kinases ERK 1/2 in rat glomerular mesangial cells. Biochem Pharmacol 2006;71:1711-9.ArticlePubMedPMC
- 47. Bagger JI, Knop FK, Holst JJ, Vilsboll T. Glucagon antagonism as a potential therapeutic target in type 2 diabetes. Diabetes Obes Metab 2011;13:965-71.ArticlePubMed
REFERENCES
Figure & Data
References
Citations
- Glucagon in type 2 diabetes: Friend or foe?
Irene Caruso, Nicola Marrano, Giuseppina Biondi, Valentina Annamaria Genchi, Rossella D'Oria, Gian Pio Sorice, Sebastio Perrini, Angelo Cignarelli, Annalisa Natalicchio, Luigi Laviola, Francesco Giorgino
Diabetes/Metabolism Research and Reviews.2023;[Epub] CrossRef
 PubReader
PubReader ePub Link
ePub Link Cite
Cite- Figure
-
- Related articles
-
- Risk Prediction and Management of Chronic Kidney Disease in People Living with Type 2 Diabetes Mellitus
- Comparative Efficacy of Rosuvastatin Monotherapy and Rosuvastatin/Ezetimibe Combination Therapy on Insulin Sensitivity and Vascular Inflammatory Response in Patients with Type 2 Diabetes Mellitus
- Comparative Effects of Sodium-Glucose Cotransporter 2 Inhibitor and Thiazolidinedione Treatment on Risk of Stroke among Patients with Type 2 Diabetes Mellitus
- Advanced Liver Fibrosis Is Associated with Chronic Kidney Disease in Patients with Type 2 Diabetes Mellitus and Nonalcoholic Fatty Liver Disease
- Association between Metabolic Syndrome and Microvascular Complications in Chinese Adults with Type 1 Diabetes Mellitus