The Search for Genetic Risk Factors of Type 2 Diabetes Mellitus
Article information
Abstract
Type 2 diabetes mellitus (T2DM) is caused by complex interplay between multiple genetic and environmental factors. The three major approaches used to identify the genetic susceptibility include candidate gene approach, familial linkage analysis and genome- wide association analysis. Recent advance in genome-wide association studies have greatly improved our understanding of the pathophysiology of T2DM. As of the end of 2010, there are more than 40 confirmed T2DM-associated genetic loci. Most of the T2DM susceptibility genes were implicated in decreased β-cell function. However, these genetic variations have a modest effect and their combination only explains less than 10% of the T2DM heritability. With the advent of the next-generation sequencing technology, we will soon identify rare variants of larger effect as well as causal variants. These advances in understanding the genetics of T2DM will lead to the development of new therapeutic and preventive strategies and individualized medicine.
INTRODUCTION
Type 2 diabetes mellitus (T2DM) is a multi-factorial disease caused by complex interplay between genetic predisposition and environmental factors [1]. Environmental factors such as increased calorie intake, physical inactivity or obesity certainly contribute to the recent diabetes epidemic. However, genetic factors are also key determinants of the individual susceptibility to T2DM. The importance of genetic risk factors for T2DM is supported by two major findings. First, there are ethnic differences in the prevalence of T2DM. Asians or Pima Indians residing in Western countries have at least a two-fold increased risk of T2DM compared to European natives [2]. Second, there is strong family history of T2DM. The offspring of T2DM parents have 40% chance of having T2DM, which is a 6-fold increased risk compared to a population risk of 7% [2].
There has been a tremendous effort to reveal the genetic predisposition of T2DM over the past 30 years or so. The three approaches adopted for identifying genetic risk factors include: 1) focusing on linkage peaks from family studies, 2) targeting candidate genes on the biological basis, and 3) genome-wide association analysis. Since the recent advent of genome-wide association studies (GWAS), there has been remarkable progress in our understanding of the genetic basis of T2DM. More than 40 genetic variations that modify the risk of T2DM development have been identified. In this article, I will review the current approaches and progress in understanding the genetics of T2DM.
FAMILY-BASED LINKAGE ANALYSIS
Family-based linkage analysis relies on genetic markers in a family pedigree to identify the chromosomal regions showing linkage with T2DM. This approach is most useful when the disease under investigation follows a monogenic form of inheritance. Causative genetic variations of several monogenic forms of diabetes, including maturity-onset diabetes of the young, neonatal diabetes and maternally inherited diabetes and deafness were successfully identified using this approach [3-5]. However, this approach failed to identify causative genes for the common form of T2DM. There have been several reports suggesting linkage peaks near CAPN10 and ACRP30 [6,7]. Unfortunately, these results were not consistent across the study population and no high-risk variation was found to be associated with T2DM near these regions.
THE CANDIDATE GENE APPROACH
The candidate gene approach refers to case-control association studies focusing on specific candidate gene or a selected genetic region, chosen based on known biological function. More than hundreds of candidate genes have been investigated by this approach. However, only a few genes such as PPARG and KCNJ11 have been shown to be associated with T2DM [8,9].
The PPARG gene encodes the peroxisome proliferator-activated receptor γ, which plays a fundamental role in adipogenesis and insulin sensitivity by regulating transcriptional activity of various genes. A variation with proline at the 12th amino acid (P12A) was confirmed to be associated with a modest (odds ratio [OR] of 1.25), but significant increase in T2DM risk [8].
The KCNJ11 gene, located on the short arm of chromosome 11, encodes the pore-forming subunit of the ATP-sensitive potassium channel Kir6.2 of the pancreatic β-cells. Gain-of-function mutations of KCNJ11 open the potassium channel and inhibit the depolarization of β-cells, leading to a defect in insulin secretion [10]. Studies in various populations have consistently reported that substitution of lysine for glutamic acid at the 23rd amino acid (E23K) is associated with an increased risk of T2DM [11,12]. In recent reports of large-scale association studies and meta-analyses, the E23K variation was found to increase the risk of T2DM with an OR of 1.15 [9].
The most significant finding derived from the candidate gene approach is the strong association of TCF7L2 gene [13]. This gene was first discovered to increase the risk of T2DM through an effort to pinpoint the previously reported linkage peak [14]. The association between rs7903146T, an intronic variation, with T2DM was replicated in almost all ethnic groups and revealed to have the strongest effect in Europeans (OR, 1.46) [15-17]. A global meta-analysis showed that this variation had an OR of 1.45 and P-value of 5.4×10-140 in a comparison of more than 46,000 cases and controls [18]. However, this allele had a significantly lower allele frequency in Asians and the resultant association was much weaker [19]. The functional role of TCF7L2 in the pathogenesis of T2DM is currently under thorough investigation. It is a crucial component of Wnt signaling and is implicated in β-cell proliferation and insulin secretion [20,21]. Furthermore, a recent study reported that TCF7L2 is important in maintaining the incretin effect [22].
GWAS
In the year 2007, there was a major breakthrough in identifying genetic risk factors of T2DM through the completion of GWAS. In GWAS, hundreds of thousands of single-nucleotide polymorphisms (SNPs) are tested for association with a disease, such as T2DM, in hundreds to thousands of individuals [23]. The three major advances that enabled this approach were 1) the improved knowledge of human genetic variations through the International HapMap project, 2) the technical advances in microarray genotyping methods, and 3) the progress in developing biostatistic methods to handle the large amount of data being produced. From the first six GWAS results reported in 2007, more than ten new genetic loci were reported to modify the risk of T2DM with genome-wide significance in Europeans [24-29]. These included variations in or near CDKAL1, CDKN2A/2B, SLC30A8, IGF2BP2, HHEX, and FTO. The variation in TCF7L2 was confirmed to have the strongest association in Europeans. In addition, the known association between variations in KCNJ11 and PPARG with T2DM were also replicated.
Based on the first GWAS reported by Sladek et al. [24], variations in SLC30A8 and near HHEX were found to be significantly associated with T2DM. Genetic variation of SLC30A8 is located in 8q24 and encodes an islet-specific zinc membrane transporter (ZnT8), which takes part in insulin synthesis and secretion [24]. Interestingly, the variation in SLC30A8, rs-13266634, results in a non-synonymous mutation of the protein [24]. One locus at 10q23-25 with large linkage disequilibrium block encompassing HHEX, IDE, and KIF11 were also significantly associated with T2DM. We have reported that variations in IDE, which encodes insulin degrading enzyme, are associated with the risk of T2DM in Koreans, as well as in a meta-analysis [30]. From the DGI study and FUSION study, variations in IGF2BP2 and CDKN2A/2B were found to be significantly associated with T2DM [25,27]. The IGF2BP2 gene located at 3q28, encodes insulin-like growth factor 2 mRNA-binding protein, which is thought to be involved in insulin signaling. A genetic variation located at 9p21 between CDKN2A and CDKN2B genes was also associated with T2DM. CDKN2A encodes p16INK4a and its overexpression results in decreased β-cell mass in senescent mice [31]. Another variation near this locus was independently associated with the risk of coronary heart disease due to elevated low density lipoprotein cholesterol concentration [32,33]. A genetic variation located in 6p22.3 of the CDKAL1 gene was first discovered to be significantly associated with T2DM through the deCODE study [28]. CDKAL1 encodes CDK5 regulatory subunit-associated protein 1-like 1 [28], which is thought to inhibit cyclin-dependent kinase 5 (CDK5) activity by binding to the CDK5 activator p35 [34]. In a recent report, disruption of CDKAL1 in mouse β-cells resulted in impaired first-phase insulin secretion [35]. Subjects who had risk variants of CDKAL1 had decreased insulin secretion capacity.
After the initial GWAS results, meta-analyses with large-scale replication analyses were performed [36,37]. The DIAGRAM consortium, which includes more than 50,000 cases and controls, was able to identify six additional genetic loci associated with T2DM [36]. These included variations in or near JAZF1, CDC123-CDMK1D, TSPAN8-LGR5, THADA, ADAMTS9, NOTCH2 genes. It should be noted that GWAS carried out in Asians revealed new T2DM genetic loci that were not previously reported in Europeans [38-40]. These were in or near KCNQ1, UBE2E2 and C2CD4A/4B. The KCNQ1 gene encodes a subunit of a voltage-gated potassium channel that is expressed in β-cells. The variation in KCNQ1 is thought to modulate the risk of T2DM by inducing β-cell dysfunction [38,39]. The UBE2E2 gene encodes the ubiquitin-conjugating enzyme E2E, which plays an important role in insulin synthesis and secretion under conditions where endoplasmic reticulum stress is increased in β-cells [40]. In an effort to find common genetic variations affecting fasting plasma glucose, variation in MTNR1B was found to be significantly associated with elevated fasting glucose and risk of T2DM [41-43]. MTNR1B encodes the melatonin receptor 2 (MT2) and it is expressed in human pancreatic β-cells as well as in the brain [42]. Increased expression of MT2 in β-cells is thought to suppress glucose-stimulated insulin secretion [44]. As of the end of 2010, the results from the DIAGRAM+ study, with a sample size of 141,000, included an additional 12 genetic variations, adding up to a total of more than 40 confirmed T2DM genetic risk loci [37].
GENETICS OF T2DM IN KOREANS
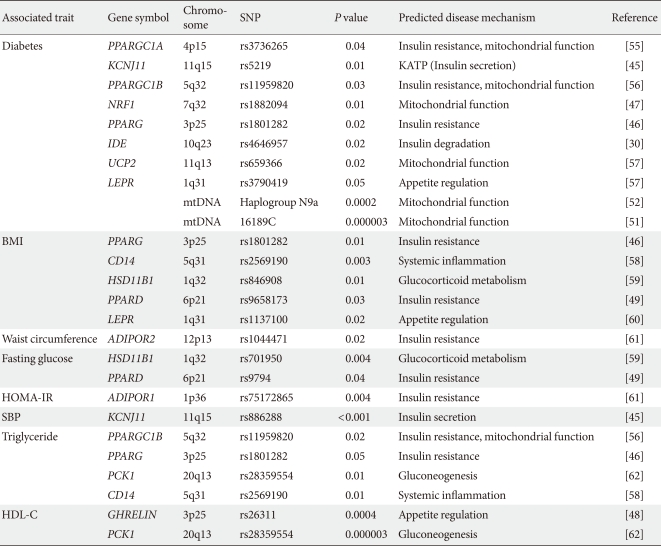
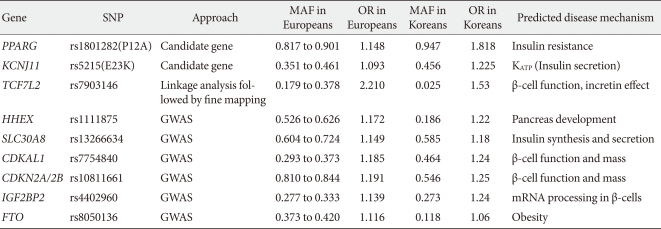
During the past ten years, my colleagues and I have focused on the candidate gene approach to find susceptibility genes for T2DM in Koreans. From these efforts, I have reported that variations in many genes, e.g., KCNJ11, PPARG, NRF1 and IDE are associated with T2DM (Table 1) [30,45-47]. We also have investigated the association of candidate genetic variations with quantitative metabolic traits such as fasting glucose, blood pressure, body mass index and dyslipidemia (Table 1) [45,48,49]. In addition, we have shown that mtDNA 16189T>C variation is associated with an increased risk of T2DM in Asians [50,51]. Among the mtDNA haplogroups, we showed that mitochondrial haplogroup N9a was significantly associated with resistance against T2DM, while D5 was significantly associated with a risk for T2DM [52]. Association of mito-chondrial DNA variation with T2DM was not observed in a European population [53,54]. For those genes identified by GWAS in Caucasians, we also confirmed the significant association between these gene variants with T2DM in Koreans (Table 2). Interestingly, the frequencies of risk alleles in Koreans were quite different from those of Caucasians, although the odds ratios of each gene variant were similar to those reported in Caucasians. These results suggest that there are important, but different contributions from genetic variants for T2DM between Caucasians and Asians (or Koreans). Fig. 1 summarizes the susceptibility genes of T2DM in the Korean population.
Genetic variations associated with T2DM and related phenotypes in Koreans
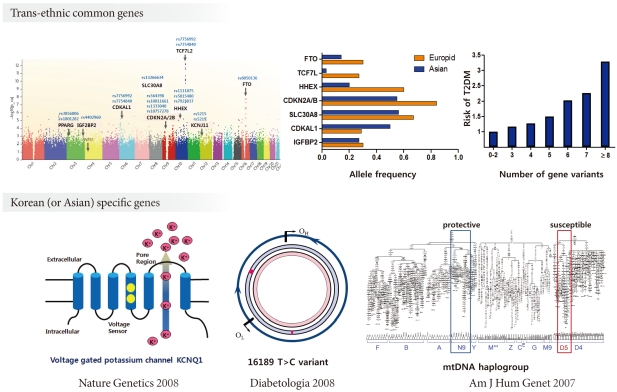
FUTURE PERSPECTIVES
It is evident that the success of the GWAS approach has revolutionized our understanding of the genetic risk factors of T2DM and has provided us with better insight into the pathogenesis of T2DM. Most of the genes identified from the initial T2DM GWAS were implicated in a defect in β-cell function, rather than increased insulin resistance. It should be noted that most genetic variations only have a modest effect and their combination only explains less than 10% of the T2DM heritability [23]. The underlying hypothesis based on the current GWAS and large-scale association studies is that common genetic variations are the cause of common complex disorders. Most of the common variations with a higher than 5% frequency reside in introns or inter-genic areas and they have a relatively small effect size.
The current and future strategies to identify T2DM risk loci include performing even larger GWAS in different ethnicities. This will lead to an increase in the number of common variations that are associated with T2DM, although the size of their effects is small. Finding rare variations with larger effect size is a rational strategy to discover additional T2DM genes. Deep sequencing around the GWAS signal might yield multiple rare variations that have functional consequences. With the advent of next generation sequencing technologies, it is now possible to perform whole exome sequencing. This will allow us to identify rare functional variation with large impact on a whole exome scale. Family studies will be used more frequently as rare variants will be enriched in the relatives of the index case and the affected status would be segregated by the causative genetic variation.
Although the progress in understanding the genetics of T2DM has already been immense, it seems that this is just the beginning of a new era. Further improvements in our understanding of T2DM genetics will eventually lead us to the development of new therapeutic and preventive methods as well as the basis for individualized medicine.
ACKNOWLEDGMENT
The author declares no conflict of interest.
Notes
The Sulwon Award for Scientific Achievement is the Korean Diabetes Association's highest scientific award and honors an individual who has excellently contributed to the progress in the field of diabetes and metabolism. Sulwon award is named after an emeritus professor Eung Jin Kim, who founded Korean Diabetes Association.
Prof. Kyong Soo Park received the first Sulwon Award at 35th Autumn Congress of Korean Diabetes Association, Nov 19-21, 2009 at Jeju, Korea.