Heterogeneity of Islet Cells during Embryogenesis and Differentiation
Article information

Abstract
Diabetes is caused by insufficient insulin secretion due to β-cell dysfunction and/or β-cell loss. Therefore, the restoration of functional β-cells by the induction of β-cell differentiation from embryonic stem (ES) and induced-pluripotent stem (iPS) cells, or from somatic non-β-cells, may be a promising curative therapy. To establish an efficient and feasible method for generating functional insulin-producing cells, comprehensive knowledge of pancreas development and β-cell differentiation, including the mechanisms driving cell fate decisions and endocrine cell maturation is crucial. Recent advances in single-cell RNA sequencing (scRNA-seq) technologies have opened a new era in pancreas development and diabetes research, leading to clarification of the detailed transcriptomes of individual insulin-producing cells. Such extensive high-resolution data enables the inference of developmental trajectories during cell transitions and gene regulatory networks. Additionally, advancements in stem cell research have not only enabled their immediate clinical application, but also has made it possible to observe the genetic dynamics of human cell development and maturation in a dish. In this review, we provide an overview of the heterogeneity of islet cells during embryogenesis and differentiation as demonstrated by scRNA-seq studies on the developing and adult pancreata, with implications for the future application of regenerative medicine for diabetes.
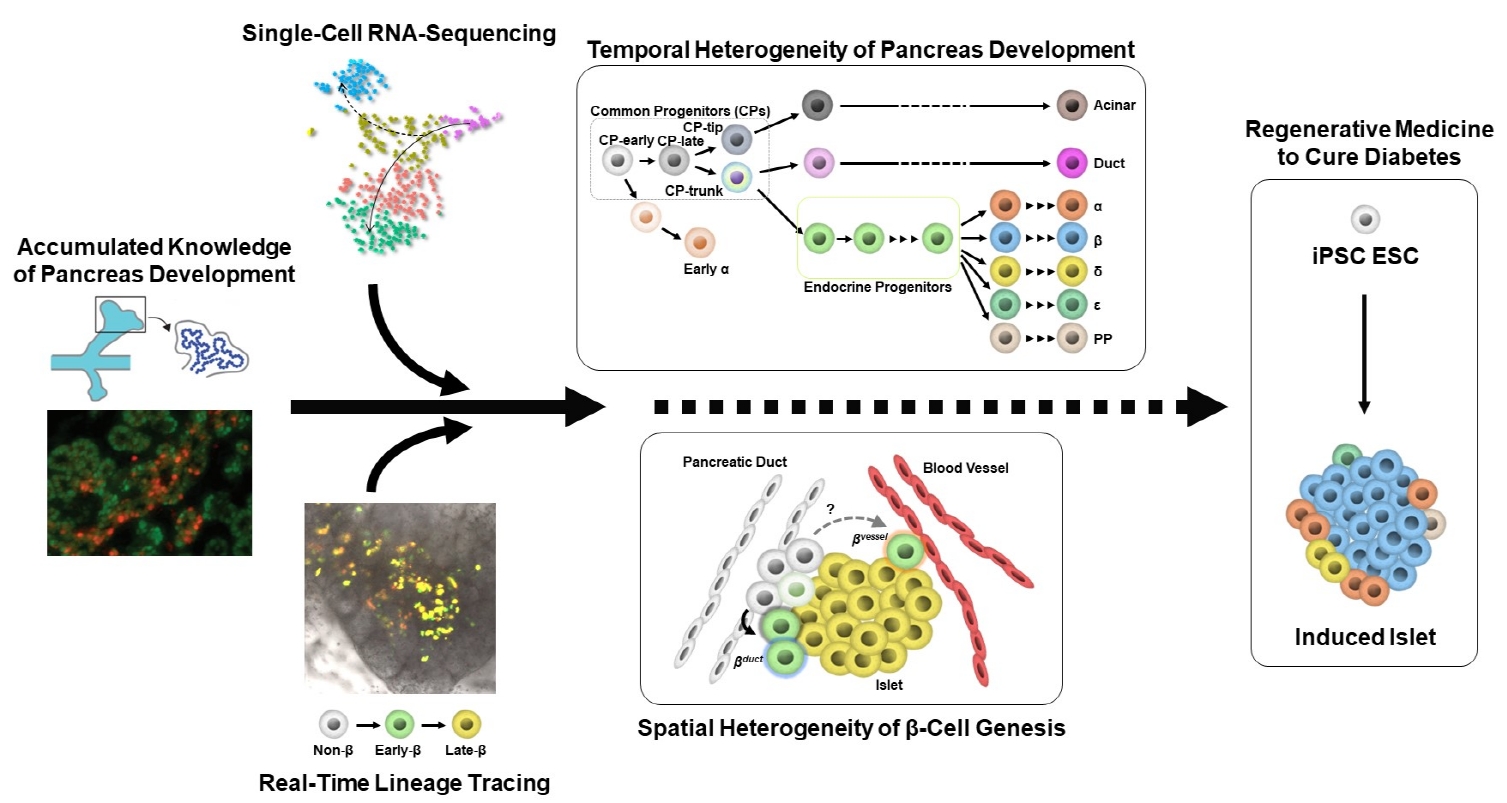
INTRODUCTION
The number of people with diabetes has been increasing globally. Diabetes affects more than 537 million people worldwide, and remains one of the most important challenges to overcome in the 21st century [1]. Poor control of diabetes leads to various complications, such as cardiovascular disease, stroke, blindness, and renal failure, resulting in a shorter lifespan and reduced quality of life [2]. One of the major pathologies in diabetes is the deficiency of insulin, which is secreted from pancreatic β-cells. Patients with type 1 diabetes mellitus (T1DM) require insulin therapy throughout their lives because most of their β-cells are destroyed by autoimmune attack. In type 2 diabetes mellitus as well as T1DM, insulin insufficiency causes glucose intolerance from an early stage of the disease [3]. Histological observations have demonstrated reduced β-cell mass [4-7]. Therefore, it is important to develop sustainable therapies to maintain and restore the number of β-cells themselves, particularly in Asia, where the population is aging, and hence patients will require long-term treatment.
One promising cell therapy for diabetes is the replenishment of β-cells by the induction of β-cell differentiation from human embryonic stem (ES) and human induced-pluripotent stem (iPS) cells. Clinical trials are underway in North America, in which ES cell-derived pancreatic endocrine progenitor (EP) cells or β-like cells are encapsulated and transplanted into T1DM patients. Various laboratories around the world are trying to develop feasible and efficient methods of inducing β-cells; however, even using the latest protocols, these β-like insulin-producing cells derived from ES or iPS cells are not mature enough to achieve glucose-stimulated insulin secretion at levels comparable to human β-cells. In addition, the presence of undesired cells, including undifferentiated cells and non-pancreatic lineage cells, can lead to tumorigenesis, which is one of the biggest safety concerns of cell transplantation. Therefore, it is essential to deeply understand the gap between surrogate β-cells and endogenous β-cells, so that the cellular differentiation into truly functional insulin-producing cells can be properly induced for the cure of diabetes.
Recently, single-cell RNA sequencing (scRNA-seq) analysis has been developed and widely used to investigate gene expression profiles at the single-cell level. This technique comprehensively detects the type and number of transcripts (mRNA) in each cell, and makes it possible to classify cell populations based on gene expression profiles. It is also possible to detect expression changes in rare cells that are difficult to detect by conventional RNA-seq analysis (which measures population averages). Furthermore, cell lineages during development and reprogramming can be traced in detail, and genes that alter their expression along the lineage can also be identified. In the field of islet biology, cell-type characteristics and heterogeneity have been reported in the human pancreas [8-10], pancreatic EPs derived from human ES cells [11], and mouse endocrine cells [12-15]. In this review, we describe the heterogeneity of islet cells during embryogenesis and differentiation, which have been clarified using cutting-edge technologies.
PANCREAS DEVELOPMENT
During embryogenesis, the pancreas develops from the definitive gut endoderm. Pancreatic specification occurs in the duodenal loop at the junction between the foregut and midgut [16]. At embryonic day 9.5 to 10 (E9.5–E10) in mice (4 to 5 weeks post-conception [wpc] in humans), the “primary transition” begins with pancreatic primordia budding on the dorsal and ventral sides of the duodenal loop. At E9.5, glucagon-producing cells (early α) first appear in the dorsal bud, followed by insulin-producing cells at E10.5, although these early hormone-producing cells probably do not contribute to the formation of mature islets [17-20].
Several key transcription factors are known to determine each developing cell type during pancreas development. The expression of pancreatic and duodenal homeobox 1 (PDX1) [21,22], SRY-box transcription factor 9 (SOX9) [23,24], and pancreas associated transcription factor 1A (PTF1A) [22,25,26] labels the multipotent common progenitors (CPs) within the pancreatic buds. These CPs give rise to the major pancreatic cell types; acinar, duct, and endocrine cells. At E11 to 12 in mice (6 wpc in humans), the two pancreatic buds (ventral and dorsal) start to bulge. The ventral bud turns to the opposite side and eventually fuses with the dorsal bud [27]. At E13.5 in mice (10 to 14 wpc in humans), this fused pancreatic bud expands with branching morphogenesis, establishing the “trunk” and “tip” domains. This event marks the start of the “secondary transition,” characterized by a rapid expansion of insulin-expressing cells. PTF1A+ and PDX1+ cells in the tip domain are initially multipotent, and subsequently become biased towards an acinar fate during the secondary transition [28]. The trunk domain contains bipotent progenitors (CP-trunk) that are positive for NK6 homeobox 1 (NKX6.1) in addition to PDX1 and SOX9, and generate duct-like structures and EPs [29]. The first somatostatin-producing cells are detected around this stage, whereas pancreatic polypeptide (PP)-producing cells appear shortly before birth [17-19].
EPs arise from the trunk domain cells lacking SOX9 and expressing neurogenin 3 (NEUROG3). NEUROG3 is necessary and sufficient for endocrine cell lineage specification [30-33], and activates downstream transcription factors that are essential for endocrine specification, including regulatory factor X6 (RFX6), neuronal differentiation 1 (NEUROD1), and paired box 4 (PAX4). In mice, NEUROG3 is expressed in a biphasic manner and correlates with the “first” and “second” transitions. The first wave of NEUROG3 expression is associated with the emergence of early α cells, between E8.5 and E11 [34,35]. The second wave of high NEUROG3 expression, between E13.5 and E18.5, leads to the generation of individual endocrine cell types [30,36,37]. In humans, NEUROG3 expression increases at 6 to 8 wpc, and peaks at 8 to 10 wpc [38]. NEUROG3+ EPs give rise to all five types of pancreatic endocrine cells (α-, β-, δ-, ϵ-, and PP-cells). Each of these endocrine cell types synthesizes and secretes one hormone, i.e., glucagon (α-cells), insulin (β-cells), somatostatin (δ-cells), ghrelin (ϵ-cells), and PP (PP-cells), although multihormonal cells are observed in both humans and mice [39-41]. The specification of these cell types is regulated by the orchestrated expression of transcription factors that trigger islet cell type-specific gene regulatory networks, and represses alternative networks to maintain cell identities [20,33,42].
HETEROGENEITY OF ENDOCRINE CELL DEVELOPMENT
Rapid advances in scRNA-seq technology have deepened our understanding of pancreas development, and have enabled this field to flourish. It is now clear that pancreatic lineage cells at various developmental stages have more heterogeneous transcriptomes than previously considered, and the subpopulations of progenitor cells are biased toward certain cell types (Fig. 1). Recently, scRNA-seq analysis of pancreata from mice at E9.5 to E17.5 revealed three consecutive subpopulations of Pdx1+ Sox9+ CPs; i.e., CP-early, CP-late, and CP cells in the tip region (CP-tip) [15]. CP-early cells expressing nuclear receptor subfamily 2 group F member 2 (Nr2f2), but not Ptf1a, emerge at E9.5, followed by the appearance of Ptf1a-expressing, but Nr2f2-negative cells (CP-late cells) at E10.5. CP-early cells had not been reported previously, and were identified as a direct source of the first wave of Neurog3+ cells, which develop into early α−cells [15]. This finding is consistent with a previous report that Ptf1a+ cells rarely contribute to endocrine lineage cells at E9.5, demonstrated by lineage tracing and single-cell quantitative polymerase chain reaction [43]. The CP-late cluster gives rise to tip-like progenitors expressing both carboxypeptidase A1 (Cpa1) and Sox9. Cpa1+ cells remain multipotent until E12.5 to E13.5, as demonstrated by lineage tracing studies, yielding trunk-duct and acinar cells [28], whereas they are restricted to the acinar lineage after E13.5 [15,44,45].
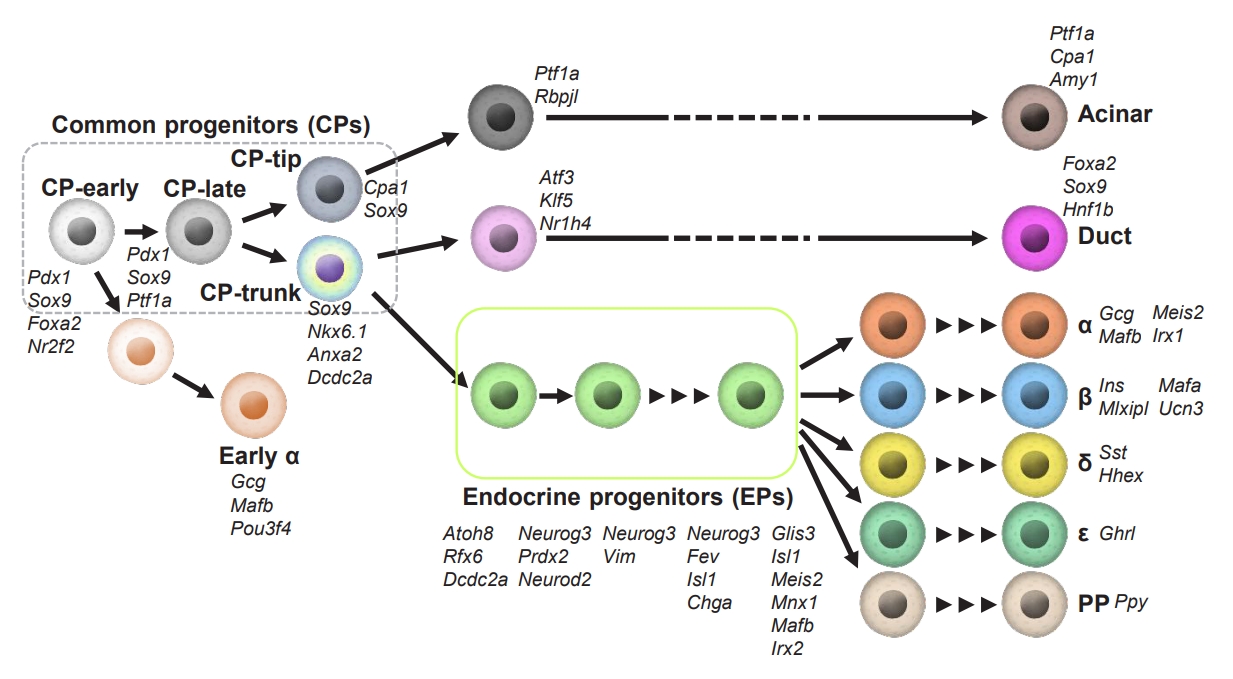
Heterogeneous properties of pancreatic cells during development. Cell-type transitions during pancreas development in mice that have been clarified by single-cell transcriptome analyses are shown. Cell-specific markers are listed near the cell type. The black arrow indicates the developmental trajectory. CP, common progenitors; EP, endocrine progenitors. Pdx1, pancreatic and duodenal homeobox 1; Sox9, SRY-box transcription factor 9; Foxa2, forkhead box protein A2; Nr2f2, nuclear receptor subfamily 2 group F member 2; Ptf1a, pancreas associated transcription factor 1A; Cpa1, carboxypeptidase A1; Nkx6.1, NK6 homeobox 1; Dcdc2a, doublecortin domain containing 2a; Gcg, glucagon; Mafb, MAF bZIP transcription factor B; Pou3f4, POU class 3 homeobox 4; Rbpjl, recombination signal binding protein for immunoglobulin kappa J region like; Amy1, amylase 1, salivary; Atf3, activating transcription factor 3; Klf5, KLF transcription factor 5; Nr1h4, nuclear receptor subfamily 1 group H member 4; Hnf1b, HNF1 homeobox B; Atoh8, atonal bHLH transcription factor 8; Rfx6, regulatory factor X6; Neurog3, neurogenin 3; Prdx2, peroxiredoxin 2; Neurod2, neurogenic differentiation 2; Vim, vimentin; Isl1, ISL LIM homeobox 1; Chga, chromogranin A; Glis3, GLIS family zinc finger 3; Meis2, Meis homeobox 2; Mnx1, motor neuron and pancreas homeobox 1; Irx2, iroquois homeobox 2; Ins, insulin; Mlxipl, MLX interacting protein like; Ucn3, urocortin 3; Sst, somatostatin; Hhex, hematopoietically expressed homeobox; Ghrl, ghrelin and obestatin prepropeptide; PP, pancreatic polypeptide.
Transcriptome analyses by scRNA-seq identified new markers for CP-trunk cells, such as annexin A2 (Anxa2) [15] and doublecortin domain containing 2a (Dcdc2a) [46], in addition to conventional markers, such as Sox9 and Nkx6.1. CP-trunk cells arise between E11.5 and E12.5, and bifurcate at E14.5 into either ductal- or endocrine-lineage cells. Interestingly, early EP cells that have already been expressing low levels of Neurog3 [15] develop into two distinct lineages at E16.5 [46]. The existence of different types of CP-trunk cells between E14.5 and E16.5 may reflect a “bias” toward either an α- or a β-cell fate. If this bias is further confirmed and the detailed mechanisms are investigated in future studies, the specification of the endocrine lineage before Neurog3 expression might be clarified. Scavuzzo et al. [46] found that angiomotin-like protein 2 (Amotl2), which is expressed in a similar pattern to Neurog3 [45-47], is expressed at a higher level in pro-β-cells at E16.5 than in pro-α cells at E14.5 in CP-trunk cells. Amotl2, which is a Hippo pathway component, inhibits the Notch and canonical Wnt pathways, induces the loss of polarity, and promotes endothelial cell motility [48-51]. The Notch and canonical Wnt pathways are known to block EP specification [52-55]. Thus, Amotl2 might regulate EP specification upstream of Neurog3 [56]. AMOTL2 knockdown in human ES cell-derived CP cells increases glucagon and decreases insulin expression at the endocrine cell stage [46]. These findings suggest that α- or β-cell priming might occur in AMOTL2-expressing CP-trunk cells. Differences in Amotl2 expression levels in the two types of CP-trunk cells between E14.5 and E16.5 may reflect the spatial heterogeneity of CP cells during delamination.
During the secondary transition at E13.5 to E17.5, EP cells switch to different cell types expressing endocrine hormones, whereas various subpopulations still coexist, as demonstrated by scRNA-seq analyses [15,45,46,57]. Low expression levels of Neurog3 were observed within the tip as well as CP-trunk cells and ductal clusters, suggesting that EPs emerge in different domains in the pancreas. The early Neurog3-low EPs are likely to reflect proliferating, long-lived cells [58,59]. The lineage tracing approach indicated that a part of these Neurog3-low EP cells acquire a ductal or acinar fate [59]. This is consistent with a recent epigenomics study through assay for transposase-accessible chromatin by sequencing (ATAC-seq) in Neurog3-deficient embryos, showing similarities in chromatin accessibility between Neurog3-deficient EPs and duct cells [60]. In the EP cells at later stages, the expression levels of Neurog3 are gradually decreased, whereas the transcription factor Fev starts to be expressed [15,37,45-47,57]. The Fev-expressing cells are confirmed in the fetal pancreas, human ES cell-derived EPs, and immature endocrine cells [10,14,61,62]. The Fev+ late EPs display an upregulation of endocrine genes, including chromogranin A (Chga), ISL LIM homeobox 1 (Isl1), iroquois homeobox 2 (Irx2), and MAF bZIP transcription factor B (Mafb). Fev+ cells may, therefore, represent an intermediate cell state between Neurog3+ EPs and more differentiated endocrine cells.
EP cells are continually being differentiated from CP cells during the secondary transition, with different development potentials between different embryonic stages [45,46]. For example, it has been shown that EP cells isolated at E14.5 have different properties from those isolated at E16.5, in the context of their transcriptome and epigenome [46]. This heterogeneity reflects the bias of E14.5 EPs toward α-cells, whereas E16.5 EPs preferentially differentiate into β-cells. Such bias of EPs are supported by a previous report that the exogenous expression of Neurog3 in Pdx1+ progenitor cells in Neurog3-null mice resulted in an age-dependent shift from pro-α to pro-β bias [63]. It has recently been reported that the direct target genes of NEUROG3 in the human stem-cell derived EPs, revealed by Cleavage Under Targets and Release Using Nuclease (CUT&RUN) technique, included not only transcription factors important for islet cell development, but also those crucial for insulin secretion in β-cells, suggesting that NEUROG3 plays a role in the bias of EPs toward β-cells [64].
Recent advances in scRNA-seq approaches further identified several markers that are specifically expressed in subpopulations of endocrine lineage cells. For example, it has been suggested that paternally expressed 10 (Peg10) and G protein subunit gamma 12 (Gng12), which are expressed in Fev+ cells at E14.5, could characterize pro-α- and pro-β-cells, respectively [57,65]. Myt1 is also identified as a marker for β-cell specification [66].
Histological analyses have suggested that EPs “delaminate” from the epithelial layer, migrate into the surrounding mesenchyme, then aggregate to form endocrine clusters, which are the so-called islets [43]. However, a recent study proposed that EPs do not fully delaminate from the epithelial cord, but instead form budding peninsula-like structures [67]. Pro-α-cells are generated first, and locate at the peninsula border, followed by pro-β-cells, which bud into the interior of the peninsula. This model may be able to explain the final architecture of mouse and small human islets. A previous scRNA-seq study detected small transient early Neurog3+ EPs at E14.5, which strongly expressed epithelial-to-mesenchymal (EMT) genes, including vimentin (Vim), possibly reflecting the delamination process [46]. We also found that somatostatin (Sst)-expressing cells, which are in an intermediate state during β-cell differentiation in both mice and humans, express high levels of Vim [65]. These EMT markers are lost in late EPs and hormone-expressing endocrine cells [46,67]. Expression of the epithelial marker E-cadherin transiently decreases but does not completely disappear [67,68]. This suggests that EPs delaminate under an EMT-like program, which partially controls peninsula-like formation.
HETEROGENEITY OF MATURING AND ADULT BETA CELLS
Single-cell approaches during embryogenesis provide the information on the transcriptional trajectories of developing endocrine cells, by applying “pseudotime” analysis. This analysis was developed based on the hypothesis that the transcriptional profiles of cells gradually and sequentially transition, although there is no precise information as to how fast a cell is differentiating on the trajectories. Even by serial scRNA-seq analyses of multiple developmental time points, mosaicism of endocrine cells at each embryonic stage makes it difficult to decipher the cell fate decisions of single cells in real time. To address this, we combined the scRNA-seq technique with a dual-color lineage tracing system so that the heterogeneity of the transcriptional pattern of single cells could be precisely investigated at a certain “real-time” points [65].
We generated “insulin-timer” and “Ins1-GFP; timer” mice by utilizing a unique feature of “timer fluorescent protein (DsRed-E5),” which shifts its fluorescence over time, and thus enables the labeling of newly generated β-cells with high time resolution [65,69,70]. This reporter system revealed that, as expected, some newborn β-cells arise close to the ductal region (βduct cells); and unexpectedly, all other β-cells arise at a location away from the ducts and adjacent to the blood vessels (βvessel cells) (Fig. 2). No newly generated β-cells were detected inside the islet-like clusters, suggesting that the β-cell-cluster (islet) forms from the periphery, consistent with the recently reported peninsula theory [67]. As Neurog3-expressing EPs emerge from the ductal region and give rise to hormone-expressing endocrine cells [31,36], it is not surprising to see Neurog3-expressing cells differentiate into βduct cells near the ductal compartment without further migration. The existence of βvessel cells suggests that a subpopulation of EPs migrates away from the ductal area toward the region near the blood vessels prior to β-cell genesis. Another possibility is that βvessel cells emerge from Neurog3-expressing EPs in the tip domains, which are recently discovered as described above [46]. Whereas several mouse models have been developed to study the regulatory mechanisms of islet cell migration [71,72], it remains unclearas to when and how β-cells are specified during migration. The unbiased scRNA-seq data with ‘Ins1-GFP; timer’ embryos demonstrated five distinct populations among newborn β-cells, confirming spatial heterogeneity of β-cell genesis, such as a high probability of glucagon (Gcg)+ βduct, MafB+ βduct, and MafA+ βvessel cells. Intriguingly, scRNA-seq of both ‘Ins1-GFP; timer’ embryos and human ES cell-derived β-like cells identified Sst-expressing cells as an earlier cluster, showing similarity in Sst-expressing cells during β-cell genesis between mice and humans. As pseudotime analyses in both models demonstrated that Sst-expressing cells are located prior to more differentiated β-cells, which express high levels of Pdx1 or Ins2 mRNAs, Sst-expressing cells are thought to be in an intermediate state during β-cell differentiation in both mice and humans. This is supported by recent reports that human embryonic stem cell (hESC)-derived β-like cells coexpress SST transcripts during differentiation [11], and by the characterization of a duct-resident somatostatin-positive EP population as a source of β-cells in homeostasis and during diabetes, as demonstrated by lineage tracing and scRNA-seq experiments [73]. Recently, Ppy-lineage cells were found to contribute to the four major types of endocrine cells, including β-cells [74]. Ppy-lineage β-cells demonstrated distinct characteristics regarding their functionality and gene expression profiles; i.e., they had reduced glucose-stimulated Ca2+ signaling response and were increased in number in experimental diabetes models. Thus, homeostatic regulation of endocrine-cell specification progresses in a heterogeneous manner.
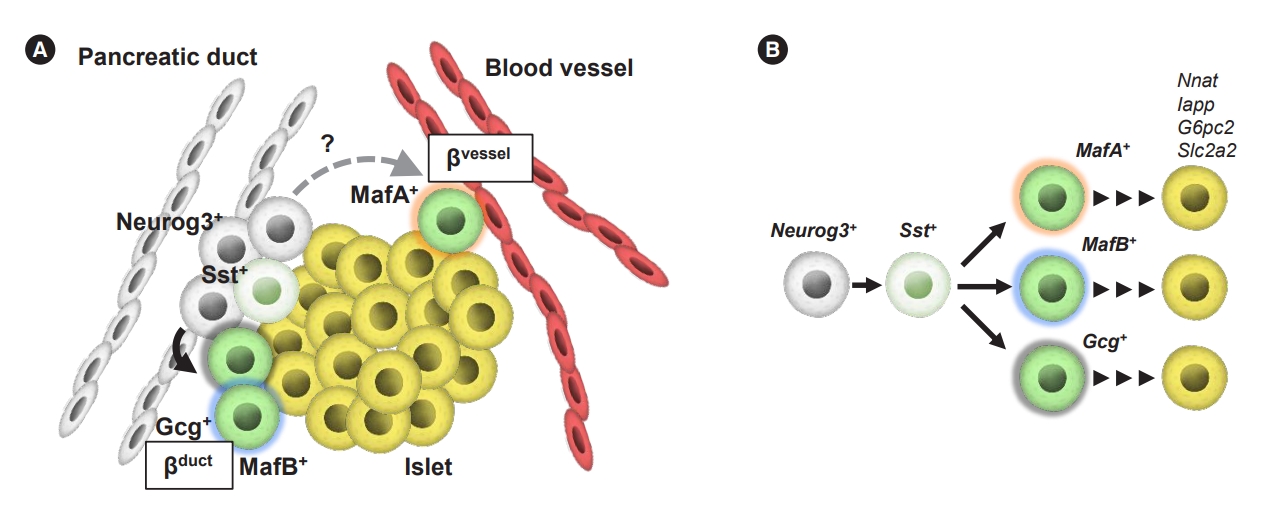
Heterogeneity of newly generated β-cells surrounded by a heterogenous spatial environment. (A) Proposed model of β-cell genesis. Endocrine progenitors differentiate into two types of β-cells; newborn β-cells near the ductal regions (βduct) and newborn β-cells adjacent to blood vessels (βvessel) cells. The βduct cells are observed in the ductal region where neurogenin 3 (Neurog3)-expressing endocrine progenitors emerge. In contrast, a subpopulation of endocrine progenitors migrates away from the ductal area toward the region near the blood vessels, and differentiate into βvessel cells. (B) Cell-type transitions in mouse β-cell development are shown. Cell-specific markers are listed near the cell type. The black arrows indicate the developmental trajectory. MafA, MAF bZIP transcription factor A; Sst, somatostatin; Gcg, glucagon; MafB, MAF bZIP transcription factor B; Nnat, neuronatin; Iapp, islet amyloid polypeptide; G6pc2, glucose-6-phosphatase catalytic subunit 2; Slc2a2, solute carrier family 2 member 2.
A series of scRNA-seq analyses of the pancreata from E17.5 embryos to postnatal day 60 (P60) adult mice were performed, focusing on endocrine cell differentiation of β-cells and α-cells using Ins1 and Gcg reporter strains, respectively [12,13]. Generally, the maturation of both β- and α-cells has been observed together with the loss of known immature endocrine markers, the expression of mature endocrine markers, and a decrease in the proportion of proliferative cells. However, these two cell types appear to have different maturation dynamics. Whereas β-cells mature steadily and gradually over time, a small population of immature β-cells remain in adult mice, demonstrated by both lineage tracing and scRNA-seq [75,76]. In contrast, all postnatal α-cells (P9–P60) cluster together indistinguishably, separately from E17.5–P0 α cells. The heterogeneity of human endocrine cells was also analyzed by scRNA-seq with human islets from different ages (newborns, toddlers, adolescents, and adults), which demonstrated that β-cells undergo age-dependent maturation, whereas α-cells retain immature characteristics to adulthood [77].
By real-time scRNA-seq analysis of late embryonic mouse β-cells, we found that early differentiated β-cells have a period of high expression of genes associated with fatty acid metabolism, such as ATP binding cassette subfamily D member 3 (Abcd3), carnitine O-octanoyltransferase (Crot), hydroxyacylCoA dehydrogenase (Hadh), ATP citrate lyase (Acly), and MLX interacting protein like (Mlxipl) [65]. This suggests that intracellular lipid energy metabolism plays an important role in early β-cells during pancreas development. The markers of immature β-cells include retinol-binding protein 4 (Rbp4) [12,13], which is also abundant in β-cell subpopulations in the adult human pancreas [9,78,79]. A combination of single-cell patch-clamp electrophysiology and scRNA-seq demonstrated that β-cells with high expression levels of RBP4 have reduced functionality, represented by lower levels of Na+ channel activity and exocytosis [79]. The expression levels of Rbp4 mRNAs were also found to be high in surviving β-cells in streptozotocin-induced diabetic mice [76]. Taken together, these findings suggest that β-cells may dedifferentiate into Rbp4-expressing cells during β-cell failure. The characteristics of maturing β-cells include a decrease in mechanistic target of rapamycin complex 1 (mTORC1) signaling and proliferative capacity [13,80,81], suppressed amino acid metabolism [13], decreased mitochondrial activity [13,80,81], and reduced production of reactive oxidative species (ROS) [13].
Adult β-cells are now widely known to be heterogeneous, although it is not clear as to how β-cell heterogeneity in adults is associated with that in embryos and infants. We will now briefly overview adult β-cell heterogeneity. Using the single-molecule fluorescent in situ hybridization (smFISH) technique, researchers identified “extreme β-cells” [82], which express extremely high levels of Ins2 mRNA, together with high mRNA levels of Pdx1, glucose transporter 2 (Glut2), ATP binding cassette subfamily C member 8 (Abcc8), etc., in the fasted state. Ins2 mRNAs and ribosomal RNAs were highly localized on the apical side, where levels of insulin protein expression were relatively low, suggesting that the extreme β-cells under fasting conditions may be tuned for basal insulin secretion, which shows temporal and functional heterogeneity in response to glucose perturbations. In terms of functionality, not all β-cells secrete insulin in a similar manner. Analysis of human islets by the reverse hemolytic plaque assay demonstrated that only 20% of the β-cell population secretes more than 90% and 70% of the total amount of insulin released at low and high glucose concentrations, respectively [83]. The observation of human islets without dissociation demonstrated that adjacent β-cells secreted insulin in synchrony with each other, suggesting that the role of β-cells differs depending on their location [84]. Furthermore, a unique optogenetic approach demonstrated that a small percentage (1% to 10%) of β-cells regulate glucose-stimulated insulin secretion as a pacemaker-like β-cell population (hub cells) [85]. In addition, β-cell subpopulations with higher glucose responsiveness have been reported, such as flattop (Fltp)+[75], CD9–/ST8 alpha-N-acetyl-neuraminide alpha-2,8- sialyltransferase 1 (ST8SIA1)– [86], ectonucleoside triphosphate diphosphohydrolase 3 (ENTPD3)+ [87-89], and protocadherin 7 (PCDH7)+ cells [90]. Although many researchers have tried to detect and quantify β-cell genesis in adult islets, it has rarely been detected in the pancreas of adult mice, indicating that β-cell mass is maintained almost exclusively by self-renewal [65,69,91]. On the other hand, rare pancreatic-cell populations have also been identified by scRNA-seq. PDX1+/activin receptor-like kinase 3 (ALK3)+/carbonic anhydrase II (CAII)– progenitors were recently identified as multipotent cells in adult human ducts [92]. An unbiased scRNA-seq approach with adult mouse pancreata identified protein C receptor-positive (Procr+) cells [93], which do not express differentiation markers and feature EMT transition characteristics that are similar to those of E14.5 EPs [46,57]. Genetic lineage tracing demonstrated that adult Procr+ islet cells undergo clonal expansion and generate all four endocrine cell types (α-, β-, δ-, and PP-cells), showing the potency of Procr+ cells as EPs in adult mouse islets.
NICHE OF THE DEVELOPING PANCREAS
During embryogenesis, specification of the pancreatic endoderm is mediated by a combination of mesoderm-derived signals, such as transforming growth factor β (TGFβ) superfamily members, retinoic acid, and fibroblast growth factors (FGFs) [94-96]. These signals synergistically drive pancreas development, regulating the expansion and differentiation of progenitors. In the chick embryo, evagination of the dorsal bud is regulated by activin-associated signals from the notochord, which is derived from the mesoderm, and represses sonic hedgehog expression, thereby leading to Pdx1 expression [97,98]. Moreover, aortic endothelial cells, which also originate from the mesoderm, are required for dorsal specification of the Pdx1-expressing endoderm, inducing the pancreas-specific transcription factor Ptf1a at least partly through Fgf10 signaling in the overlying dorsal mesenchyme [99-102]. It has been reported that ectopic vascularization leads to ectopic insulin expression [99]. Furthermore, the close interaction between β-cells and endothelial cells was demonstrated to play a role in insulin gene expression and β-cell proliferation [103]. In our recent study, the presence of βvessel cells suggested a role of vascular endothelial cells in β-cell specification [65]. As βvessel cells express Mafa, which is a transcription factor that has been shown to be expressed in mature β-cells, βvessel cells may develop more mature features than βduct cells, even though the temporal window after activation of the Ins1 promoter should be equivalent. Direct signals from surrounding tissues, including vascular endothelial cells and nerve cells, and/or substances carried by blood vessels, such as nutrients and oxygen, are likely to affect the heterogeneity of endocrine-cell maturation. Further studies are needed to address the microenvironment around βduct and βvessel cells, which may lead to the discovery of new molecules that are crucial for β-cell specification and maturation.
In addition to the importance of endothelial cells in pancreatic endocrine development, mesenchymal cells have been shown to be required for pancreas development [104]. The mesenchyme plays various developmental roles at different timepoints [105,106]. Studies involving scRNA-seq have not only focused on the pancreatic epithelium, but have also shed light on mesenchymal subclusters, demonstrating their heterogeneity at E12.5 to E18.5 [14,46,57]. In the E14.5 mouse pancreas, several mesenchymal subpopulations were identified, in which unique secreted factors associated with TGFβ, Hippo, and inhibitor of DNA-binding (ID) signaling pathways, are enriched. Another mesenchymal cluster in the E12.5 to E14.5 pancreas was identified as being composed of NKX2.5-expressing cells, which are splenopancreatic mesenchymal cells surrounding the pancreatic buds [106,107]. These findings emphasize the importance of the mesenchymal niche around the developing pancreas in endocrine differentiation. Identifying essential signals and morphogens from pancreatic mesenchymal cells will help us to establish a safe and efficient method for generating stem cell-derived β-cells.
CONCLUSIONS
In this article, we overviewed the heterogeneity of islet cells during pancreas development, as revealed by scRNA-seq and other approaches, which repeatedly demonstrated that pancreatic β-cells are a collection of insulin-expressing cells with similar but different characteristics. As we gain a better understanding of cell populations at the single-cell and high-resolution level, we will be able to clarify (1) the detailed molecular basis during the differentiation of immature β-cell subtypes into mature cells, (2) the change in compositional ratios of β-cell subtypes in diabetes, and (3) a possible method to artificially control β-cell heterogeneity. With the rapid progress of multi-omics analysis and imaging technology at the single-cell level, it may not be long before these questions are clarified. In addition to the widely used scRNA-seq and scATAC-seq, new single-cell technologies, including single-cell CUT&Tag [108], proteomics [109], and metabolomics [110] will bring us to a new dimension. Single-cell clustered regularly interspaced short palindromic repeats (CRISPR) screens, such as Perturb-seq, are promising tools for genotype-phenotype mapping, which provide pooled information on genetic perturbations and associated multivariate phenotypic changes in individual cells [111]. On the other hand, it is necessary to carefully determine whether and to what extent the heterogeneity of rodent β-cells can be translated into that of human β-cells. By combining the accumulated knowledge in developmental biology with the recent data from stem cell research, we believe that it will become possible in the near future to develop regenerative medicine techniques that can cure diabetes.
Notes
CONFLICTS OF INTEREST
No potential conflict of interest relevant to this article was reported.
FUNDING
None
Acknowledgements
None