Epidemiology, Pathophysiology, Diagnosis and Treatment of Heart Failure in Diabetes
Article information

Abstract
The cardiovascular disease continuum begins with risk factors such as diabetes mellitus (DM), progresses to vasculopathy and myocardial dysfunction, and finally ends with cardiovascular death. Diabetes is associated with a 2- to 4-fold increased risk for heart failure (HF). Moreover, HF patients with DM have a worse prognosis than those without DM. Diabetes can cause myocardial ischemia via micro- and macrovasculopathy and can directly exert deleterious effects on the myocardium. Hyperglycemia, hyperinsulinemia, and insulin resistance can cause alterations in vascular homeostasis. Then, reduced nitric oxide and increased reactive oxygen species levels favor inflammation leading to atherothrombotic progression and myocardial dysfunction. The classification, diagnosis, and treatment of HF for a patient with and without DM remain the same. Until now, drugs targeting neurohumoral and metabolic pathways improved mortality and morbidity in HF with reduced ejection fraction (HFrEF). Therefore, all HFrEF patients should receive guideline-directed medical therapy. By contrast, drugs modulating neurohumoral activity did not improve survival in HF with preserved ejection fraction (HFpEF) patients. Trials investigating whether sodium-glucose cotransporter-2 inhibitors are effective in HFpEF are on-going. This review will summarize the epidemiology, pathophysiology, and treatment of HF in diabetes.
INTRODUCTION
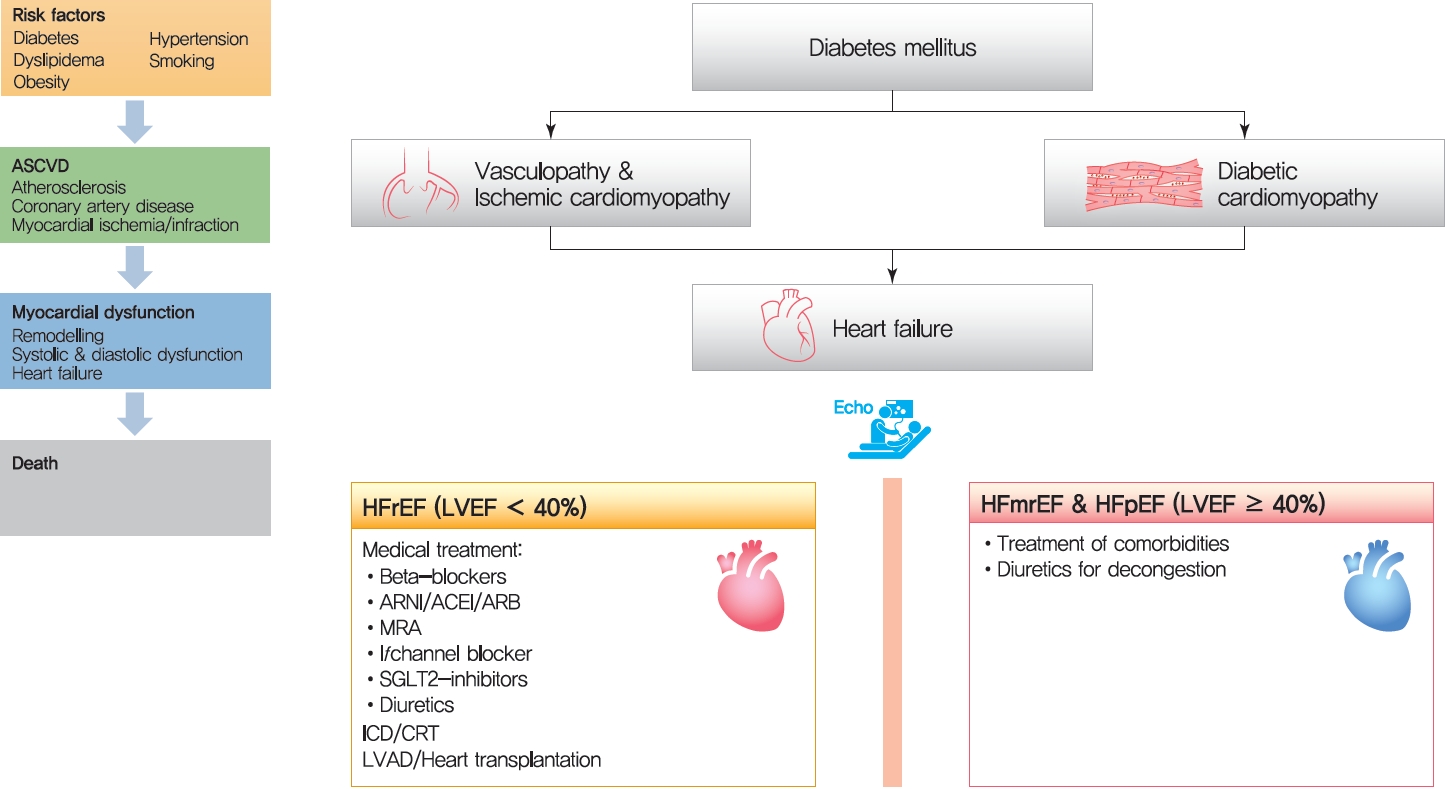
Thirty years ago, Dzau and Braunwald [1] introduced the cardiovascular disease continuum and framed cardiovascular disease as a chain of events, initiated by a myriad of related and unrelated risk factors and progressing through numerous physiological pathways and processes to the development of end-stage heart disease (Fig. 1) [2]. The cardiovascular disease continuum begins with diabetes mellitus (DM), hypertension, and dyslipidemia, among others, then results in advanced heart failure (HF) and cardiovascular death. The cardiovascular disease continuum also emphasizes the possibility that therapeutic intervention at every stage may prevent or slow the development of symptomatic HF and hopefully prolong life [2]. However, this concept has been untrue for diabetes because clinical trials showed that intensive glucose-lowering therapy did not translate into better clinical outcomes in diabetic patients [3-6].

The cardiovascular disease continuum. ASCVD, atherosclerotic cardiovascular disease.
"Updated on 30 September 2021"
Not all diabetic patients develop HF, and not all HF patients have diabetes [7,8]. Nonetheless, diabetes is an important risk factor for the development of HF. Diabetes increases the risk for HF [9-11] and complicates its course, such that HF patients with DM had worse outcomes than those without DM [12,13].
The scientific interest in HF in DM has increased significantly with the publications of recent cardiovascular outcome trials of newer antidiabetic drugs that robustly showed clinical benefit and altered the cardiovascular disease continuum for the first time [14,15]. This review will summarize the epidemiology, pathophysiology, and management of HF in diabetes from the viewpoint of a clinical cardiologist specialized in HF management.
EPIDEMIOLOGY OF DM AND HF
Diabetes is a serious and increasing global health burden. The number of people with diabetes increased from 108 million in 1980 to 422 million in 2014, in which 8.5% of adults ≥18 years had diabetes. It is expected that over 592 million people worldwide will have diabetes by 2035. Regarding death, 1.6 million deaths were directly caused by diabetes in 2016 [16,17].
According to the diabetes fact sheet in Korea 2020, the prevalence of diabetes among adults 30 years or older was 13.8%, representing approximately 4.94 million Koreans in 2020. The prevalence was 27.6% among adults aged ≥65 years. Approximately 61.3% of patients with DM have coexisting hypertension, and in patients aged ≥65 years with DM, 74.3% also had hypertension. DM and hypertension are independent risk factors for HF, and their coexistence predisposes excess cumulative risk for HF development [18].
Diabetes is associated with a 2- to 4-fold increased risk of HF. In the Framingham Heart Study, DM was associated with a nearly 2-fold increase in the risk of incident HF in men and a 4-fold increase in women, even after adjustment for other cardiovascular risk factors [10]. In patients with known coronary artery disease in the Heart and Soul Study, DM was also associated with a 3.3-fold higher adjusted risk of incident HF [19]. Hence, DM is an important risk factor that promotes the progression of each stage in the cardiovascular disease continuum. Other risk factors for incident HF include older age, longer diabetes duration, ischemic heart disease, greater body weight, and higher creatinine level, among others; therefore,patients with multiple risk factors should receive special medical attention [20].
Among HF patients, the prevalence of DM is 2 to 2.5 times higher than in the general population. In the Korean Heart Failure (KorHF) registry from 1998 to 2003, the prevalence of DM was 31% [21]. In the Korean Acute Heart Failure Registry (KorAHF) from 2004 to 2009, 36% had diabetes [7]. These are similar to the prevalence of DM in Europe (33%) in the EuroHeart Failure Survey II (EHFS II) [22] but found to be less than that of the United States, with 44% according to the Acute Decompensated Heart Failure National Registry (ADHERE) [23]. Interestingly, only 11% of African HF patients had diabetes in the Sub-Saharan Africa Survey of Heart Failure (THESUS-HF) registry [24].
HF patients with DM had more unfavorable characteristics compared to those without DM. They have more ischemic etiology, higher body mass index, heart rate, systolic blood pressure, creatinine level, and N-terminal pro-B-type natriuretic peptide level than those without DM, which may explain the worse clinical outcomes of these patients [25].
PATHOPHYSIOLOGY
The exact pathophysiologic mechanism linking diabetes and HF is unknown. Hyperglycemia, insulin resistance, and hyperinsulinemia all seem to initiate and perpetuate disease progression. Diabetes can cause myocardial ischemia via vasculopathy (both micro- and macrovasculopathy) and can directly exert deleterious effects on the myocardium (cardiac myocyte and interstitium).
Vasculopathy
Prolonged exposure to hyperglycemia causes vasculopathy, and there exists a linear relationship between glucose level and its detrimental effects. The concept of the “glycemic continuum” reflects that the negative effects of hyperglycemia occur even at levels below the threshold for DM diagnosis [26,27]. Both micro- and macrovasculopathy are the principal causes of morbidity and mortality in diabetic patients. Regarding myocardial perfusion, DM causes clinical and subclinical ischemia via both macro- and microvasculopathy, leading to myocardial dysfunction.
Pathophysiology of diabetic macrovasculopathy
In the center of vasculopathy, the alteration of endothelial and vascular smooth muscle cells (VSMCs) plays a key role. Under normal conditions, the endothelium constitutively produces nitric oxide (NO) via endothelial NO synthase [28]. NO causes vasodilatation via activation of guanylyl cyclase in VSMCs. It also inhibits the proliferation and migration of VSMCs, thus inhibiting the atherosclerotic process [29-31]. In contrast, the loss of NO increases pro-inflammatory activity via activation of nuclear factor kappa B (NF-κΒ); expression of leukocyte adhesion molecules and production of chemokines and cytokines [31], which promotes monocyte and VSMC migration into the intima and formation of macrophage foam cells.
The bioavailability of NO is determined by its synthase and degradation. Its level also reflects vascular health. In patients with diabetes, hyperglycemia, free fatty acids [32-34], and insulin resistance [35,36], increased reactive oxygen species (ROS) activate protein kinase C, leading to low NO levels [37-39] and endothelial dysfunction. These alterations in vascular homeostasis due to endothelial and VSMC dysfunction favor a pro-inflammatory/thrombotic state, which ultimately leads to atherothrombosis.
Pathophysiology of diabetic microvasculopathy
At the level of the capillaries, abnormal intercellular signaling in endothelial cells decreases the capillary diameter and induces microvascular rarefaction in diabetic conditions in a human in vitro model of angiogenesis and in mice [40,41]. In diabetic porcine models, DM caused alterations in capillary structures at 2 months after DM induction and myocardial hypoperfusion before the development of significant epicardial coronary artery stenosis (Supplementary Fig. 1) [42].
Impairment of myocardial perfusion
Coronary circulation consists of epicardial coronary arteries, arterioles, and capillaries. Myocardial perfusion occurs during the diastole of each cardiac cycle following the pressure gradient between the epicardial coronary artery and left ventricular (LV) diastolic pressure; the distribution of blood flow then matches the dynamic needs of local tissue metabolism, which is regulated by arterioles and capillaries [43,44]. Because myocardial oxygen extraction is near-maximal at rest, myocardial oxygen delivery is almost dependent on coronary blood flow. In diabetic patients, accelerated atherosclerosis with luminal narrowing in the epicardial coronary artery was believed to be the main mechanism for the insufficient blood supply. However, many diabetic patients have “paradoxical” absence of significant stenosis in epicardial arteries with significant ischemic symptoms and positive exercise test [45-47], implying coronary microvasculopathy [48,49].
The coronary flow reserve (CFR) is the ratio between hyperemic and resting coronary flow, and the reduction of CFR has been reported to represent coronary microvascular dysfunction [50]. A reduction in CFR and myocardial blood flow was associated with increased cardiac mortality in diabetic patients without coronary artery stenosis [51-54].
Vasculopathy-independent myocardial dysfunction
Diabetic cardiomyopathy is defined by the existence of abnormal myocardial structure and performance in the absence of other cardiac risk factors, such as coronary artery disease, hypertension, and significant valvular disease, in individuals with DM [55]. Although the exact mechanisms need to be elucidated, several mechanisms have been proposed for the development of diabetic cardiomyopathy.
Hyperglycemia causes the formation of advanced glycation end products (AGEs), which are glycated proteins or lipids after prolonged exposure to glucose. AGEs can cross-link with extracellular matrix proteins, increase fibrosis, and impair myocardial relaxation [56,57]. AGEs can also cause intracellular damage via activation of the receptors for AGEs, leading to an increase in cytosolic ROS and activation of inflammatory pathways via NF-κΒ signaling [58,59].
ROS can mediate mitochondrial uncoupling and reduce cardiac energy efficiency [60]. Impairment in mitochondrial bioenergetics results in impaired intracellular calcium handling. Calcium reuptake via sarcoendoplasmic reticulum calcium transport ATPase (SERCA)-2 into the sarcoplasmic reticulum is an energy-dependent process and may result in both contraction and relaxation abnormalities [61]. Impaired glucose tolerance and increased fatty acid uptake by cardiac myocytes may exceed mitochondrial oxidative capacity, leading to lipid over-storage and production of lipotoxic metabolites and ROS (Fig. 2) [62,63].
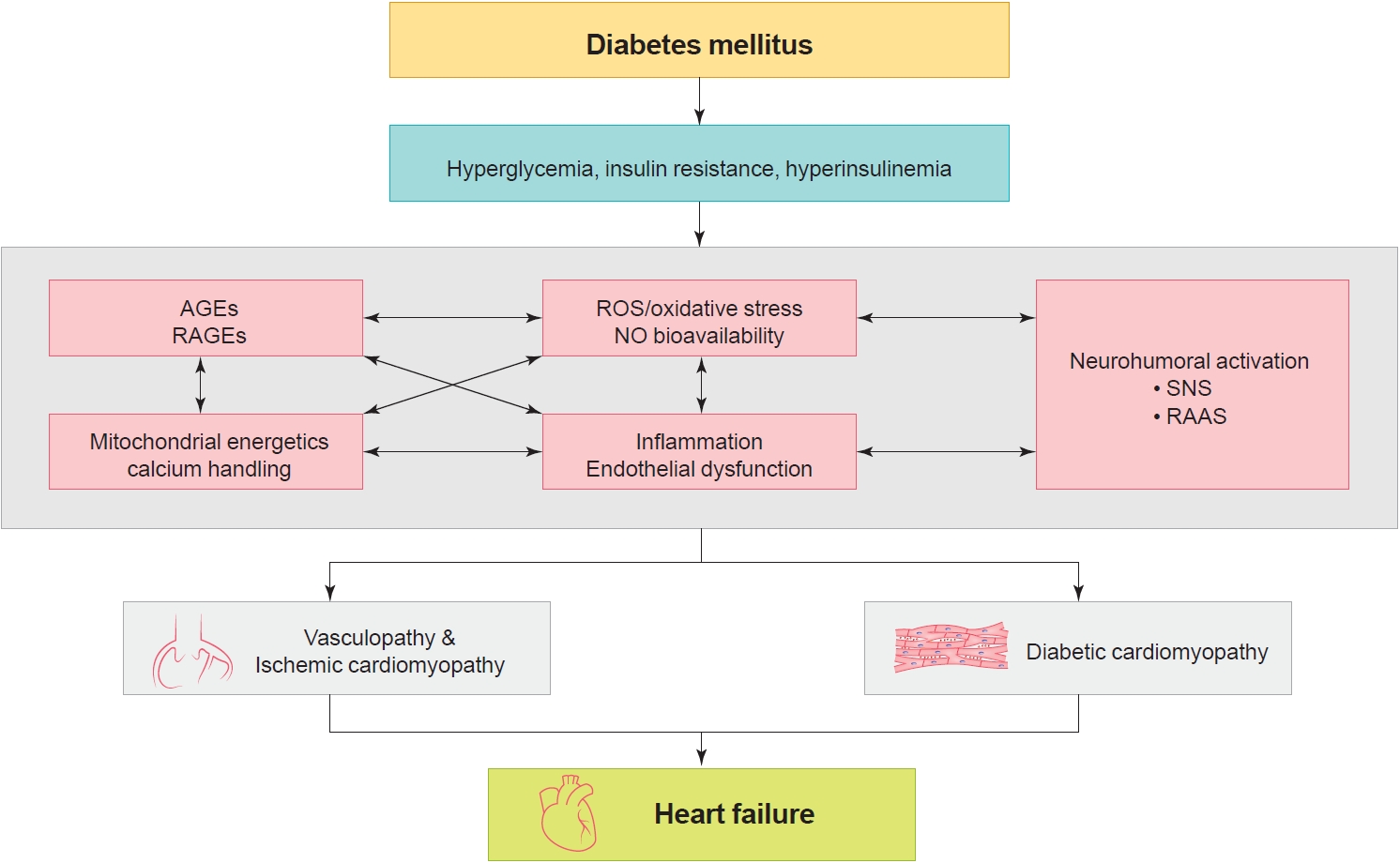
Pathophysiology for heart failure development in diabetes. AGE, advanced glycation end product; RAGE, receptors for AGE; ROS, reactive oxygen species; NO, nitric oxide; SNS, sympathetic nervous system; RAAS, renin-angiotensin-aldosterone system.
There exists a chronic activation of the renin-angiotensin-aldosterone system in diabetic patients. Increased angiotensin II (AT-II) level causes vasoconstriction and increases the afterload and promote LV hypertrophy. AT-II also promotes collagen production, extracellular matrix protein accumulation leading to myocardial contractile dysfunction [64].
Oxidative stress, inflammation, impaired mitochondrial energetics, intracellular calcium handling, and increased neurohumoral activation all contribute to the anatomic and functional alteration associated with diabetic cardiomyopathy.
DIAGNOSIS OF HEART FAILURE
For diagnosis of HF, the patients should have typical symptoms (e.g., shortness of breath, fatigue) and signs (e.g., pulmonary and ankle edema) of HF, and objective evidence of functional and structural cardiac abnormality leading to reduced cardiac output and/or increased intracardiac filling pressure [65]. Currently, HF types are further classified according to the left ventricular ejection fraction (LVEF) and defined as HF with reduced ejection fraction (HFrEF) (LVEF<40%), HF with mid-range ejection fraction (HFmrEF) (40%≤ LVEF <50%), and HF with preserved ejection fraction (HFpEF; LVEF ≥50%) [65]. For diagnosis of HFmrEF and HFpEF, the patients should additionally have elevated levels of natriuretic peptides and either relevant structural heart disease such as LV hypertrophy and/or left atrial enlargement and/or diastolic dysfunction (Fig. 3A) [65]. In the KorAHF registry, 59.1%, 15.8%, and 25.1% had HFrEF, HFmrEF, and HFpEF, respectively [66]. Among patients with diabetes 64%, 14.4%, and 21.6% had HFrEF, HFmrEF, and HFpEF, respectively.
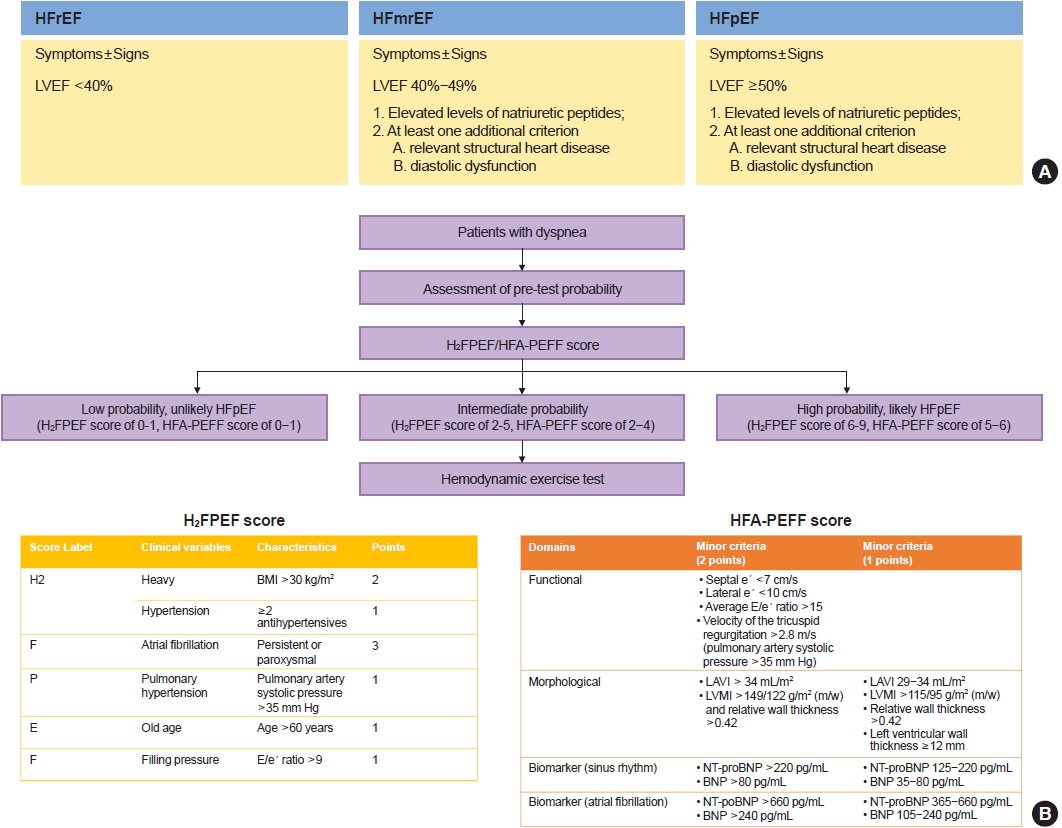
Diagnosis of heart failure. (A) Definition of heart failure types according to the ejection fraction. (B) Algorithm for the diagnosis of heart failure with preserved ejection fraction (HFpEF). HFrEF, heart failure with reduced ejection fraction; HFmrEF, heart failure with mid-range ejection fraction; LVEF, left ventricular ejection fraction; H2FPEF, Heavy (a body mass index >30 kg/m2 , 2 points), Hypertension (use of ≥2 antihypertensive medications, 1 point), atrial Fibrillation (3 points), Pulmonary hypertension (pulmonary artery systolic pressure >35 mm Hg, 1 point), Elderly (age >60 years, 1 point), and Filling pressures (E/e´ >9, 1 point); HFA-PEFF, Heart Failure Association—Pretest assessment, (ii) diagnostic workup with Echocardiogram and natriuretic peptide score, (iii) advanced workup with Functional testing in case of uncertainty, and (iv) Final etiological workup; BMI, body mass index; LAVI, left atrial volume index; LVMI, left ventricular mass index; NT-proBNP, N-terminal pro-B-type natriuretic peptide; BNP, B-type natriuretic peptide.
The diagnosis of HFpEF is challenging. Since HFpEF diagnosis based on echocardiographic data and natriuretic peptide levels has limited sensitivity, revised algorithms with scoring systems have been proposed recently.
Reddy et al. [17] proposed a H2FPEF score which consists of six dichotomized variables: Heavy (a body mass index >30 kg/m2, 2 points); Hypertension (use of ≥2 antihypertensive medications, 1 point); atrial Fibrillation (3 points); Pulmonary hypertension (pulmonary artery systolic pressure >35 mm Hg, 1 point); Elderly (age >60 years, 1 point); and Filling pressures (E/e´ >9, 1 point). The score ranges from 0 to 9 and at a score of ≥6, HFpEF was diagnosed with a probability ≥90%.
The Heart Failure Association algorithm of the ESC (HFA-PEFF) consists of (1) Pretest assessment, (2) diagnostic workup with Echocardiogram and natriuretic peptide score, (3) advanced workup with Functional testing in case of uncertainty, and (4) Final etiological workup [67]. In the calculation of the HPA-HEFF score, for each major criterion met, 2 points are awarded, whereas 1 point is awarded for a minor criterion. A score of ≥5 based on echocardiographic and natriuretic peptide levels is diagnostic of HFpEF. A score of ≤1 makes a diagnosis of HFpEF very unlikely.
Borlaug [68] proposed a practical approach to the diagnosis of HFpEF. After an initial clinical assessment of the patient’s history, symptoms, and signs, the risk score is calculated. Additional workup in the form of invasive and non-invasive diastolic stress test is recommended for patients with intermediate probability to confirm the HFpEF diagnosis (Fig. 3B).
TREATMENT OF HEART FAILURE
HFrEF and HFpEF are characterized by different anatomy and degree of neurohumoral activation. Patients with HFrEF have an enlarged LV cavity and relatively small LV wall thickness, whereas patients with HFpEF have relatively normal LV diameter, but increased wall thickness [69]. According to the law of Laplace, wall tension correlates directly with the LV diameter and the pressure, but inversely with the wall thickness. Therefore, HFrEF patients have higher wall tension and higher natriuretic peptide levels that are secreted by ventricles in response to increased wall stress [70]. The two distinct HF types respond differently to the drugs that modulate neurohumoral activation.
Medical treatment of HFrEF
Currently, there are seven classes of drugs that improved the clinical outcomes in HFrEF and they are (1) renin-angiotensin-system (RAS) inhibitors, (2) angiotensin receptor neprilysin inhibitor (ARNI), (3) mineralocorticoid receptor antagonist (MRA), (4) beta-blockers, (5) If -channel inhibitor, (6) sodium-glucose cotransporter-2 (SGLT2) inhibitors, and (7) soluble guanylate cyclase stimulator.
In the Studies of Left Ventricular Dysfunction (SOLVD) [71], enalapril 10 mg twice daily reduced the mortality in hospitalized HF patients with LVEF ≤35% compared to placebo. In the Valsartan Heart Failure Trial (Val-HeFT) with patients with LVEF <40%, valsartan 160 mg twice daily reduced the composite endpoint of mortality and morbidity, defined as the incidence of cardiac arrest with resuscitation, hospitalization for HF (HHF), or receipt of intravenous inotropic or vasodilator therapy compared to placebo by 13% (relative risk, 0.87; 97.5% confidence interval, 0.77 to 0.97; P=0.009) [72]. However, the morality was similar in the two groups. It is of note that angiotensin receptor blockers (ARBs) are reserved for patients who cannot tolerate angiotensin-converting enzyme inhibitors (ACEis).
ARNI consists of an ARB and a neprilysin inhibitor, an endopeptidase that degrades vasoactive peptides such as natriuretic peptide, bradykinin, and adrenomedullin. A natriuretic peptide is considered the “natural antagonist” of angiotensin and has natriuretic, diuretic, vasodilatory, antifibrotic and antisympathetic effects [73]. In the Prospective Comparison of ARNI with ACEi to Determine Impact on Global Mortality and Morbidity in HF (PARADIGM-HF) study sacubitril-valsartan 200 mg twice daily reduced the composite of cardiovascular deaths and HHF by 20% (hazard ratio, 0.80; 95% confidence interval, 0.73 to 0.87; P<0.001) compared with enalapril 10 mg twice daily in optimally treated HFrEF patients with LVEF ≤40% [74].
In the randomized aldactone evaluation study (RALES), spironolactone 25 mg daily reduced morality by 30% (relative risk of death, 0.70; 95% confidence interval, 0.60 to 0.82; P<0.001) in patients with LVEF <35%. Electrolyte should be monitored regularly because hyperkalemia can occur.
The effect of beta-blockers in HFrEF seems to be substratespecific. Currently, three beta-blockers, i.e., carvedilol [75], bisoprolol [76], and metoprolol succinate [77] showed a beneficial effect in HFrEF. It is of note that beta-blockers have not been tested in acute HF, and in some meta-analyses their effect was neutral in patients with AF [78].
Low heart rate is associated with better survival. Ivabradine blocks If channel in the sinus node and slows heart rate without exerting a negative inotropic effect. In the Systolic Heart failure treatment with the IF inhibitor ivabradine Trial (SHIFT) [79], ivabradine reduced the composite of all-cause mortality and HHF by 20% in patients with LVEF ≤35% in sinus rhythm and with a heart rate >70 beats/min. In patients with heart rate >75 beats/min ivabradine also showed survival benefit [80].
SGLT2-inhibitors block glucose reuptake in the proximal tubules. In cardiovascular outcome studies in diabetic patients with and without atherosclerotic cardiovascular disease, empagliflozin, canagliflozin, dapagliflozin, and ertugliflozin reduced all HHF [81-85]. In patients with HFrEF, both dapagliflozin and empagliflozin reduced the composite of all-cause death or HHF by approximately 25% and HHF by approximately 30% [86]. This effect was consistent in those with and without DM [25].
In VerICiguaT GlObal Study in Subjects With Heart Failure With Reduced EjectIon FrAction (VICTORIA) study [87], vericiguat, an oral soluble guanylate cyclase stimulator that improves the cardiac contractility, reduced the composite of all-cause deaths and HHF by 10% (hazard ratio, 0.90; 95% confidence interval, 0.82 to 0.98; P=0.02) in severe HF patients with LVEF <45%.
With the accumulation of clinical evidence, the algorithm for pharmacologic treatment has been changing (Fig. 4) [88]. The latest expert consensus recommends the initial use of beta-blockers and ARNI/ACEi/ARB. ARNI is preferred over ACEi or ARB. Each drug should be up titrated to the maximum tolerated dose every 2 weeks (Table 1). The addition of MRA, SGLT2-inhibitors, and ivabradine should also be considered in appropriate patients. Diuretics may be added and up titrated to achieve decongestion. Regular monitoring for renal function, electrolytes imbalance, and cardiac function is required.

Treatment algorithm for guideline-directed medical therapy. HFrEF, heart failure with reduced ejection fraction; ARNI, angiotensin receptor-neprilysin inhibitor; ACEi, angiotensin-converting enzyme inhibitor; ARB, angiotensin receptor blocker; BB, beta-blocker; eGFR, estimated glomerular filtration rate; NYHY, New York Heart Association; MRA, mineralocorticoid receptor antagonist; SGLT2, sodium-glucose cotransporter-2; HR, heart rate.
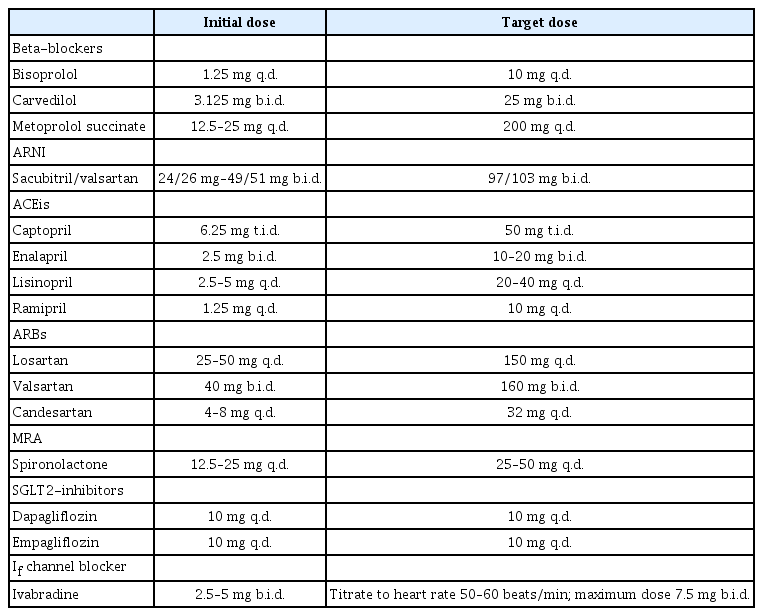
Guideline-directed medical therapy: drugs names and doses
Device therapy in HFrEF
HF patients are at increased risk for sudden cardiac death which is mainly caused by ventricular arrhythmias, bradycardia, and asystole. An implantable cardiac defibrillator (ICD) can treat bradycardia and potentially lethal ventricular arrhythmia. ICD is recommended for primary prevention in symptomatic HF patients with LVEF ≤35%, despite >3 months of optimal medical therapy (OMT).
Some HF patients show dyssynchronous LV contraction resulting ineffective translation of the LV contraction into stroke volume. Cardiac resynchronization therapy (CRT) is a modality of cardiac pacing that simultaneously paces the ventricles and restores the ventricular synchrony. It has been shown to improve symptoms and reduce morbidity and mortality. CRT is recommended for symptomatic patients with HF in sinus rhythm with a QRS duration >130 ms and with LVEF ≤35% despite OMT.
Treatment of HFpEF
Since HFmrEF patients were generally included in HFpEF trials, the treatment for HFmrEF and HFpEF are considered the same. Until now, no drugs in HFrEF improved survival in HFpEF [89-92], although some may reduce the HHF. Despite the different response to medical therapy, interestingly, HFrEF and HFpEF have a similar prognosis [93]. Cardiovascular deaths remain the main cause of mortality in HF; however, the proportion of non-cardiovascular deaths is higher in HFpEF (30% to 40%) than HFrEF (15% to 20%) [94]. Therefore, the current practice guidelines emphasize the treatment of underlying disease. In HFpEF patients with congestion, diuretics can alleviate symptoms of HF.
Regarding SGLT2-inhibitors, sotagliflozin, an SGLT1 and 2 dual blocker reduced a composite of cardiovascular deaths and hospitalizations and urgent visits for HF in patients with diabetes and recent worsening HF, regardless of LVEF in the Effect of Sotagliflozin on Cardiovascular Events in Patients With Type 2 Diabetes Post Worsening Heart Failure (SOLOIST-WHF) study [95]. However, the early termination of the trial and the small sample size of this subgroup made it difficult to draw any firm conclusion. Designated trials are on-going to evaluate the benefit of SGLT2 inhibitors in HFpEF patients [96,97].
CONCLUSIONS
Diabetes is an important risk factor for HF. Prolonged hyperglycemia, hyperinsulinemia, and insulin resistance can cause alterations in vascular homeostasis with reduced NO and increased ROS levels, which activate pro-inflammatory pathways that lead to atherothrombotic progression and myocardial dysfunction. HF patients with DM have worse prognosis than those without DM. The classification, diagnosis and treatment of HF remain the same for patients with and without DM. Until now, drugs targeting neuro-humoral and metabolic pathways improved mortality and morbidity in HFrEF, but not in HFpEF. Thus, all HFrEF patients should receive guideline-directed medical therapy.
SUPPLEMENTARY MATERIALS
Supplementary materials related to this article can be found online at https://doi.org/10.4093/dmj.2020.0282.
Vascular changes in the diabetic pigs. Optical coherence tomography images of the epicardial coronary arteries of diabetic and control pigs at 8 weeks showed no difference in lumen diameter. By contrast, diabetic pigs had a significantly smaller capillary diameter, more irregularity and disconnection. In cardiac magnet resonance imaging, diabetic pigs had lower myocardial perfusion suggesting that the microvasculopathy is the main cause for decreased myocardial perfusion at the early stages of diabetes.
Notes
CONFLICTS OF INTEREST
No potential conflict of interest relevant to this article was reported.
FUNDING
This work was supported by the SNUBH Research Fund (Grant no 14-2015-029).
Acknowledgements
None