Nonalcoholic Fatty Liver Disease: A Drug Revolution Is Coming
Article information

Abstract
The worldwide prevalence of non-alcoholic fatty liver disease is around 25%, and that of nonalcoholic steatohepatitis (NASH) ranges from 1.5% to 6.45%. Patients with NASH, especially those with fibrosis, are at higher risk for adverse outcomes such as cirrhosis and liver-related mortality. Although vitamin E, pioglitazone, and liraglutide improved liver histology in randomized trials, there are currently no Food and Drug Administration-approved drugs for NASH. Five pharmacologic agents—obeticholic acid, elafibranor, cenicriviroc, resmetirom, and aramchol—are being evaluated in large, histology-based phase 3 trials. Within 2 to 4 years, new and effective drugs for the treatment of NASH are expected. Additionally, many phase 2 trials are ongoing for various agents. Based on the results of phase 2 and 3 trials, combination treatments are also being investigated. Future treatment strategies will comprise drug combinations and precision medicine based on the different phenotypes of NASH and treatment response of the individual patient.
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) can be classified histologically into nonalcoholic fatty liver or nonalcoholic steatohepatitis (NASH). The worldwide prevalence of NAFLD is around 25%, and that of NASH ranges from 1.5% to 6.45% [1]. Patients with NASH, especially those with fibrosis, are at higher risk for adverse outcomes such as cirrhosis and liver-related mortality [2-7]. The most important histological feature of NAFLD associated with long-term mortality is fibrosis (stage 2–4) [2,8,9]. Therefore, one of the following two surrogates must be satisfied in phase 2b or 3 trials for conditional approval of drug development: (1) resolution of NASH without worsening of fibrosis or (2) a reduction in fibrosis by one or more stages without worsening of NASH [10,11]. Although there are currently no Food and Drug Administration (FDA) approved drugs for NASH, vitamin E, pioglitazone, and liraglutide improved the liver histology of patients with NASH in randomized trials [12-16]. Despite its potential benefits, vitamin E has been associated with conflicting reports of increased overall mortality [17,18], in haemorrhagic stroke [19] and prostate cancer in males older than 50 [20]. Pioglitazone causes weight gain [12-14], and its usefulness for NASH is still under investigation. Therefore, these risks must be balanced with the potential benefit in NASH patients, who have no options for treatment other than lifestyle modification. At present, weight loss and lifestyle modification with diet and exercise is recommended as the first-line therapy [21-24]. However, long-term compliance with lifestyle modification is difficult to achieve and maintain in the target population. Therefore, a major unmet need for a new drug to resolve NASH and reverse liver fibrosis exists. Recent main therapeutic targets for NASH are bile acid pathway, insulin resistance, inflammation, thyroid hormone receptor (THR)-β stimulation, hepatic lipid metabolism, anti-fibrosis, and so on. Five pharmacologic agents—obeticholic acid (OCA; farnesoid X receptor [FXR] agonist), elafibranor (a peroxisome proliferator-activated receptor [PPAR] α and δ agonist), cenicriviroc (CVC; an dual antagonist of C–C chemokine receptor [CCR] types 2 and 5), resmetriom (THR-β agonist), and aramchol (stearoyl-CoA desaturase [SCD] 1 inhibitor)—improved liver histology in phase 2 studies [25-29], and are undergoing phase 3 studies to evaluate their long-term efficacy and safety. Additionally, multiple novel agents targeting NASH-related pathways are the subjects of phase 1 and 2 trials, and approximately 200 pharmacologic agents are being evaluated for NASH treatment. This review summarizes the mechanisms of action of the main pharmacologic agents and outlines the pivotal phase 2 and 3 studies that have been completed or are ongoing.
DRUG CLASSIFICATION BASED ON THE ACTION OF MECHANISM
Bile acid pathways
Farnesoid X receptor agonist, obeticholic acid
OCA is derived from the primary human bile acid, chenodeoxycholic acid, which is a natural FXR agonist [30]. As a result of synthetic modification, OCA stimulates FXR activity 100-fold more intensely than chenodeoxycholic acid [30]. FXR is a nuclear receptor that is highly expressed in the liver and small intestine, and plays an important role in the synthesis and enterohepatic circulation of bile acids (Fig. 1) [31]. FXR activation reduces bile acid synthesis by inhibiting the conversion of cholesterol to bile acids, and it possesses antiinflammatory and antifibrogenic activity [32]. Activation of FXR in the ileum also inhibits the uptake of bile acids by downregulating the sodium-dependent bile acid transporter. Finally, OCA exerts anticholestatic and hepatoprotective effects by regulating the metabolism of cholesterol and bile acids [33].
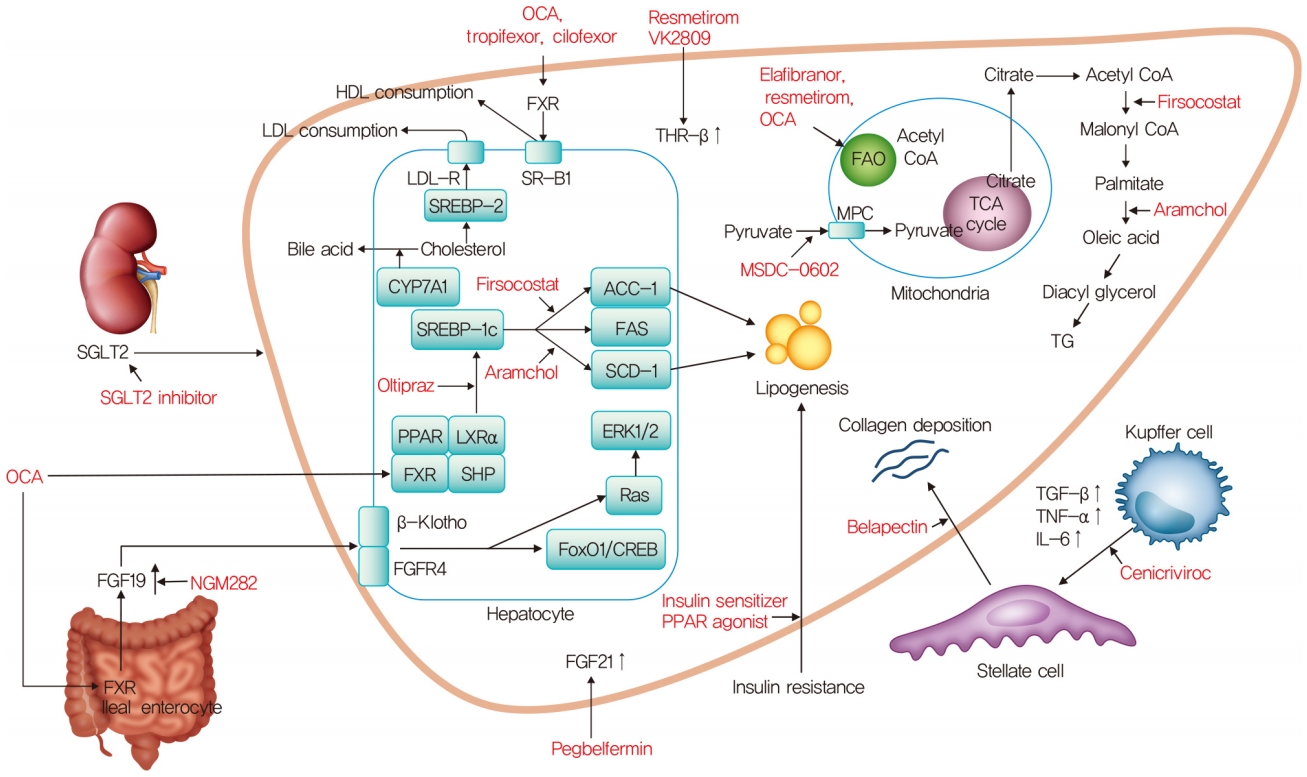
Mechanism of action for nonalcoholic steatohepatitis treatment. OCA, obeticholic acid; HDL, high density lipoprotein; LDL, low density lipoprotein; SGLT2, sodium glucose cotransporter 2; FXR, farnesoid X receptor; THR-β, thyroid hormone receptor-β; LDL-R, low density lipoprotein receptor; SR-B1, scavenger receptor class B type 1; SREBP-2, sterol regulatory element-binding proteins-2; CYP7A1, cholesterol 7α-hydroxylase; SREBP-1c, sterol regulatory element binding protein-1c; ACC-1, acetyl-coenzyme A carboxylase-1; FAS, fatty acid synthase; SCD1, stearoyl-CoA desaturase 1; PPAR, peroxisome proliferator-activated receptor; LXRα, liver X receptor α; SHP, small heterodimer partner; ERK1/2, extracellular signal-regulated kinase 1/2; FGFR4, fibroblast growth factor receptor 4; FoxO1, forkhead box protein O1; CREB, cAMP response element-binding protein; FGF, fibroblast growth factor; FAO, fatty acid β-oxidation; CoA, coenzyme A; TCA, tricarboxylic acid; MPC, mitochondrial pyruvate carrier; TG, triglyceride; TGF-β, transforming growth factor-β; TNF-α, tumor necrosis factor-α; IL-6, interleukin-6.
In a phase 2a clinical trial on OCA, patients with type 2 diabetes mellitus (T2DM) and NAFLD were randomly assigned to groups given placebo (n=23), 25 mg OCA (n=20), or 50 mg OCA (n=21) once daily for 6 weeks (NCT00501592) [34]. Insulin sensitivity increased by 24.5% (P=0.011) in the combined OCA groups, whereas it decreased by 5.5% in the placebo group. The OCA groups had significant reductions in the levels of γ-glutamyltransferase (γ-GT) and alanine aminotransferase (ALT) and dose-related weight loss. Also, the levels of markers of liver fibrosis decreased significantly in the group treated with 25 mg OCA. In a phase 2b clinical trial, non-cirrhotic patients with NASH were randomly assigned 1:1 to groups given placebo (n=142), and 25 mg OCA (n=141) once daily for 72 weeks (FLINT, NCT01265498) [25]. Fifty (45%) of 110 patients in the OCA group had improved liver histology compared with 23 (21%) of 109 patients in the placebo group (relative risk, 1.9; 95% confidence interval [CI], 1.3 to 2.8; P=0.0002). Also, 23% of the OCA group developed pruritus compared with 6% of the placebo group. OCA improved the histological features of NASH, but its long-term benefit and safety need further clarification.
In the interim analysis of an ongoing, phase 3 study of OCA, patients with NASH, an NAFLD activity score (NAS) of at least 4, and fibrosis stages F2–F3, or F1 with at least one accompanying comorbidity were randomly assigned in a 1:1:1 ratio to receive oral placebo, OCA 10 mg, or OCA 25 mg daily (GENERATE, NCT02548351) (Table 1) [35]. The primary analysis involved 931 patients with stage F2–F3 fibrosis (311 in the placebo group, 312 in the OCA 10 mg group, and 308 in the OCA 25 mg group). The fibrosis improvement endpoint was achieved by 37 (12%) patients in the placebo group, 55 (18%) in the OCA 10 mg group (P=0.045), and 71 (23%) in the OCA 25 mg group (P=0.0002). The NASH resolution endpoint was not met. However, the results of this interim analysis showed that OCA 25 mg significantly improved fibrosis and key components of NASH activity.

Ongoing phase 3 trials for patients with NASH
In the safety population (1,968 patients with fibrosis stages F1–F3) of this trial, the most common adverse event was pruritus, which occurred in 51% of the OCA 25 mg group, 28% of the OCA 10 mg group, and 19% of the placebo group. Pruritis was generally mild to moderate in severity; however, 9% of the OCA 25 mg safety population discontinued the drug because of pruritus. The lipoprotein profile was also monitored in the safety population. The mean low density lipoprotein cholesterol (LDL-C) and total cholesterol levels peaked at 3 months in the OCA group. Also, 17% of the OCA group and 7% of the placebo group needed statin treatment.
A recent phase 2 trial (CONTROL, NCT02633956) evaluating the use of OCA combined with a statin for NASH showed that use of OCA at 5, 10, or 25 mg daily increased the LDL-C level after 4 weeks of treatment, and the addition of atorvastatin 10 mg decreased the LDL-C level to below baseline in all OCA groups by week 8 [36]. The combination of OCA and atorvastatin was generally safe and well tolerated.
The scope of OCA has been expanded to cirrhosis, and a phase 3 trial of OCA in patients with compensated cirrhosis due to NASH is ongoing. Patients were randomly assigned 1:1:1 to groups given 10 mg OCA, 10 mg with titration to 25 mg OCA at 3 months, or placebo for 18 months (REVERSE, NCT03439254) (Table 1). The REVERSE study uses a dose-escalation approach as hepatic decompensation arises in patients with advanced cholestatic liver disease treated with OCA [37]. The primary endpoint is the proportion of subjects with improvement in fibrosis by at least one stage with no worsening of NASH.
Non-bile acid FXR agonist
1) Tropifexor (LJN-452)
Tropifexor (TXR; LJN452) is a highly potent, non-bile acid FXR agonist that induces the expression of target genes at very low doses without significant Takeda G-protein coupled receptor clone 5 activation [38]. A 48-week phase 2 trial on TXR in patients with NASH is ongoing (FLIGHT-FXR, NCT02855164) (Table 2) [39]. In an interim analysis after 12 weeks, similar to OCA, TXR demonstrated unfavorable lipid changes with a dose-related increase in the LDL-C level and a decrease in the high density lipoprotein cholesterol (HDL-C) level, in addition to pruritus, at higher doses compared with placebo. There was also a relative decrease in liver fat content by magnetic resonance imaging proton density fat fraction (MRI-PDFF) of −9.8% in placebo, −16.9% with TXR 60 mg, and −15.6% with TXR 90 mg, in the interim analysis. Further analysis demonstrated that TXR was more efficacious in patients with a lower body mass index (BMI), suggesting that a weight-based dosing approach may be necessary [40].

Pivotal phase 2 trials for NASH treatment
2) Cilofexor (GS-9674)
Because OCA has problematic side effects such as pruritus, hypercholesterolemia, and hepatic decompensation, selective non-bile acid synthetic FXR agonists have been developed. Cilofexor (GS-9674) is a potent, selective, nonsteroidal agonist of FXR that primarily functions to activate FXR in the intestine and does not undergo enterohepatic circulation [41]. Intestinal FXR agonism by cilofexor accentuates the physiologic release of fibroblast growth factor (FGF) 19, and may mitigate the detrimental effects of systemic FXR activation, including dyslipidemia, pruritus, and hepatotoxicity [41,42]. In a proof-of-concept study, 10 patients with NASH and F2–3 fibrosis who received 30 mg of cilofexor once daily for 12 weeks experienced decreased hepatic fat level and stiffness, and improved liver biochemistry [43].
In a phase 2 trial, 140 non-cirrhotic and NASH patients were randomized to cilofexor 100 mg (n=56), cilofexor 30 mg (n=56), or placebo (n=28) for 24 weeks (NCT02854605) (Table 2) [44]. The results indicated that 100 mg of cilofexor reduced the hepatic fat content and was reasonably well tolerated. Pruritus was not common but was more frequent in the 100 mg (14%) versus 30 mg (4%) cilofexor and placebo groups (4%). No difference in pruritus was seen at the 30 mg dose compared with placebo but, like many other FXR agonists, cilofexor caused pruritis dose dependently, with more moderate to severe pruritus in those receiving 100 mg daily compared with placebo. Cilofexor for 24 weeks resulted in significant reductions in hepatic steatosis, liver biochemistry, and serum bile acids in patients with NASH. Other FXR agonists (nidufexor [45], and EDP-305 [46]) have been developed and are in phase 2 trials.
Fibroblast growth factor 19 analogue, NGM282
The FGF family of hormones mediate metabolic functions and tissue repair and regeneration [47]. FGF19 is a downstream target of FXR activation, with FXR initiating FGF19 secretion by the intestine. FGF19 is a hormone that regulates bile acid synthesis and glucose homeostasis [48], and NGM282 is an engineered analogue of FGF19 (Fig. 1).
In a phase 2 study, 82 patients with biopsy proven NASH were randomly assigned to receive 3 mg (n=27) or 6 mg subcutaneous NGM282 (n=28) or placebo (n=27) (NCT02443116) [49]. The primary endpoint was the absolute change in liver fat content from baseline to week 12. Responders were patients who achieved a 5% or greater reduction in absolute liver fat content as measured by MRI-PDFF. At 12 weeks, 20 (74%) patients in the 3 mg dose group and 22 (79%) in the 6 mg dose group achieved at least a 5% reduction in absolute liver fat content from baseline versus two (7%) in the placebo group. NGM282 produced rapid and significant reductions in liver fat content with an acceptable safety profile in patients with NASH. In a recent open-label study, the histological efficacy of NGM282 in patients with biopsy proven NASH was assessed [50].
Paired liver biopsies from 43 patients who received subcutaneous NGM282 (1 mg, n=24; 3 mg, n=19) once daily for 12 weeks were evaluated, blinded to time point, subject, and clinical information. At week 12, NGM282 improved the histological features of NASH, with significant reductions in the NAS and fibrosis scores, accompanied by improvements in noninvasive imaging and serum markers. A larger phase 2 study with a target of 250 participants with biopsy proven NASH is currently actively recruiting (NCT02443116) (Table 2).
Pegylated fibroblast growth factor 21, pegbelfermin (BMS-986036)
FGF21 has also been implicated in bile acid pathways. Activation of FXR, together with PPARα, induces hepatic expression and secretion of FGF21 [51]. FGF21, a non-mitogenic hormone, is a key regulator of energy metabolism [52]. Endogenous FGF21 has a short half-life of 1 to 2 hours, but various modification strategies have been used to create longer-acting FGF21 analogues [53]. Pegbelfermin (BMS-986036) is a polyethylene glycol-conjugated recombinant analogue of human FGF21 with a prolonged half-life that enables up to weekly dosing.
In a randomized, double-blind, phase 2a study, 75 patients with a BMI of at least 25 kg/m², biopsy-confirmed NASH (fibrosis stage 1–3), and a hepatic fat fraction of at least 10% by MRI-PDFF were randomized into three groups (25 patients to receive 10 mg pegbelfermin once daily; 24 to receive 20 mg pegbelfermin once weekly, and 26 to receive placebo) [54]. There was a significant decrease in the absolute hepatic fat fraction in the group receiving 10 mg pegbelfermin daily and in the group receiving 20 mg pegbelfermin weekly compared with the placebo group. Most adverse events were mild; the most common was diarrhea in eight (16%) of 49 patients treated with pegbelfermin. Treatment with pegbelfermin for 16 weeks was generally well tolerated and significantly reduced the hepatic fat fraction in patients with NASH. Currently, two large phase 2 trials (FALCON 1, n=160; FALCON 2, n=152) are ongoing to assess the safety and efficacy of pegbelfermin (NCT03486899 and NCT03486912, respectively) (Table 2).
Insulin resistance
Peroxisome proliferator-activated receptor agonist
1) Elafibranor
PPARs are nuclear receptors playing key roles in cellular processes regulating metabolic homeostasis, immune-inflammation, and differentiation. There are three nuclear receptor isoforms, PPARα, PPARβ/δ, and PPARγ, which are encoded by different genes [55]. Elafibranor (GFT505) is a dual a PPARα and δ agonist, and it regulates lipid and insulin metabolism. PPARα is most prominently expressed in the liver and is activated by hypolipidemic fibrates. PPARα controls the lipid flux in the liver by modulating fatty acid transport and β-oxidation and improves plasma lipids by decreasing the triglyceride level and increasing that of HDL-C [56]. In advanced NASH, the PPARα level is reduced but recovers after improvement [57]. PPARδ (also called PPARβ) regulates metabolism in the liver and peripheral tissues. PPARδ agonists enhance fatty acid transport and oxidation, increase the HDL level, and improve glucose homeostasis by enhancing insulin sensitivity and inhibiting hepatic glucose output [58]. In a pilot trial, a selective PPARδ agonist reduced liver fat content, improved insulin sensitivity and plasma lipid levels, and decreased the γ-GT level [59].
In a phase 2b clinical trial on elafibranor, patients with NASH without cirrhosis were randomized to receive elafibranor 80 mg (n=93), elafibranor 120 mg (n=91), or placebo (n= 92) daily for 52 weeks (GOLDEN-505, NCT01694849) [26]. The primary outcome was resolution of NASH without fibrosis worsening, using protocol-defined and modified definitions. In an intention-to-treat analysis, there was no significant difference between the elafibranor and placebo groups in the primary outcome. However, in a post hoc analysis of patients with NAS ≥4 (n=234), elafibranor 120 mg resolved NASH in a larger proportion of patients than placebo based on the protocol definition (20% vs. 11%; odds ratio, 3.16; P=0.018) and the modified definitions (19% vs. 9%; odds ratio, 3.52; P=0.013). Also, patients in whom NASH resolved after receiving elafibranor 120 mg had reduced liver fibrosis compared to those without NASH resolution. The levels of liver enzymes, lipids, and markers of systemic inflammation, as well as the glucose profile, were significantly reduced in the elafibranor 120 mg group versus the placebo group. Elafibranor was well tolerated and did not cause weight gain or cardiac events, but did produce a mild, reversible increase in the serum creatinine level.
A phase 3 trial on elafibranor in 2,000 NASH patients (NAS ≥4) with stage 2/3 fibrosis is ongoing (RESOLVE-IT, NCT0270-4403) (Table 1). The primary outcome is the proportion of patients with resolution of NASH without worsening of fibrosis at 72 weeks. The trial also evaluated a composite long-term outcome composed of all-cause mortality, cirrhosis, and liver-related clinical outcomes at 4 years. The trial began in March 2016, and the results are due in December 2021.
2) Lanifibranor
Lanifibranor (IVA337) is a moderately potent agonist of all three PPAR isoforms, with well-balanced activation of PPARα and PPARδ and partial activation of PPARγ [60]. While other PPAR agonists target one or two PPAR isoforms, lanifibranor is the only pan-PPAR agonist in clinical development.
In a phase 2b clinical trial on lanifibranor (IVA337) in NASH patients with liver steatosis and moderate to severe necroinflammation without cirrhosis, 247 patients were randomly assigned to receive lanifibranor 800 mg, 1200 mg, or placebo per day for 24 weeks (NATIVE, NCT03008070) (Table 2). The primary endpoint was a decrease from baseline in the SAF (steatosis, S; activity, A; and fibrosis, F) activity score. The study began in January 2017 and was completed in March 2020. Initial results are expected in the first half of 2020.
Glucagon-like peptide-1 receptor agonist, semaglutide
No pharmacotherapies have been established for patients with NASH and T2DM. The main point is to determine the best antidiabetic drugs for treatment of NASH, to prevent the progression of hepatic fibrosis and cardiovascular or renal events. Glucagon-like peptide-1 receptor agonists (GLP-1 RAs) and sodium-glucose cotransporter 2 (SGLT2) inhibitors are expected to ameliorate NASH and NAFLD. GLP-1 is a gut-derived incretin hormone that induces weight loss and insulin sensitivity. GLP-1 analogs are approved for use in patients with T2DM and obesity. These analogs provide the benefits of weight loss, improved glycemic control, fewer hypoglycemic events, and a reduced frequency of major cardiovascular events [61,62]. There is interest in the therapeutic role of GLP-1 RAs for NAFLD and NASH [15,63-66].
In a recent randomized, placebo-controlled trial involving 52 patients with biopsy-proven NASH, 1.8 mg of liraglutide administered subcutaneously daily for 48 weeks was associated with greater resolution of NASH and less progression of fibrosis [15]. However, the American Association for the Study of Liver Diseases practice guidance 2018 recommended that it is premature to consider GLP-1 agonists for treating liver disease in patients with NAFLD or NASH [24].
Semaglutide is a novel GLP-1 analogue with an extended half-life of approximately 1 week. In a phase 2 study of the effect of semaglutide on NASH, 320 patients with NASH were randomly assigned to placebo or three dose levels of semaglutide subcutaneously for 72 weeks (NCT02970942) (Table 2). The primary endpoint was NASH resolution without worsening of fibrosis. Initial results from the study are expected in May 2020, with completion anticipated in July 2020. Another phase 2 study of semaglutide involves once-weekly injection of semaglutide, 2.4 mg, for 48 weeks. The primary outcome is improvement of liver fibrosis by at least 1 stage with no worsening of NASH after 48 weeks (NCT03987451) (Table 2). Semaglutide is the most promising of various GLP-1 analogues for the treatment of diabetic NASH. However, whether GLP-1 analogues improve hepatic inflammation or fibrosis in NASH is unknown.
Sodium-glucose cotransporter 2 inhibitor, dapagliflozin
SGLT2 inhibits glucose reabsorption in the proximal tubule, leading to glucouria and a reduction in the plasma glucose level. Therefore, SGLT2 inhibitors have therapeutic potential for NASH and NAFLD. Small studies have shown the effect of dapagliflozin in patients with NAFLD and T2DM [67,68]. Although the possibility cannot be excluded that the reduction in body weight or visceral adipose tissue caused by dapagliflozin may be associated with a decrease in liver steatosis or fibrosis, dapagliflozin showed a beneficial effect in patients with NAFLD. A multicenter, randomized, placebo-controlled, phase 3 clinical trial is ongoing to assess the efficacy and safety of dapagliflozin for treating NASH (DEAN, NCT03723252) (Table 1). The primary endpoint is improvement of the liver histological score at 12 months. One-hundred patients with NASH were randomly assigned to placebo or 10 mg of dapagliflozin. The study began in March 2019, and the results are due in November 2021.
Mitochondrial target of thiazolidinedione, MSDC-0602K
First-generation insulin-sensitizing thiazolidinediones directly bind to and activate the PPARγ nuclear hormone receptor and are used to treat T2DM [69]. However, they are associated with significant side effects including edema, bone fractures mediated by PPARγ, and hypoglycemia. The second-generation insulin sensitizer MSDC-0602K was designed to reduce these side effects, while still producing insulin sensitizing pharmacology in animal models of diabetes [70] and NASH [71]. It modulates the effects of over-nutrition at the level of the mitochondrial pyruvate carrier (MPC) with minimal PPARγ binding (Fig. 1) [72]. Initial studies showed that MSDC-0602 could increase lipid oxidation and reduce de novo lipid synthesis and gluconeogenesis in the liver, both in vivo and in vitro, without the side-effects of first-generation insulin sensitizers [73].
In a 52-week randomized, double-blind, placebo-controlled phase 2b study, patients with biopsy-confirmed NASH and fibrosis (F1–F3) were randomly assigned to placebo (n=94), or 62.5 mg (n=99), 125 mg (n=98), or 250 mg (n=101) of MSDC-0602K (EMMINENCE, NCT02784444) (Table 2) [74]. The primary efficacy endpoint was hepatic histological improvement of ≥2 points in NAS with a ≥1-point reduction in ballooning or lobular inflammation and no increase in fibrosis stage at 12 months. In that study, MSDC-0602K did not exert a significant effect on liver histology. However, MSDC-0602K significantly decreased the levels of fasting glucose, insulin, glycated hemoglobin, and markers of liver injury without dose-limiting side effects. A phase 3 study will be initiated in 2020 (MMONARCh, NCT03970031).
Inflammation
C-C chemokine receptor types 2 and 5 antagonist, cenicriviroc
CVC is an oral, dual antagonist of CCR2 and CCR5. Blockade of CCR2, a chemokine receptor predominantly expressed on monocytes and macrophages, results in reduced recruitment, migration and infiltration of these cells to the injured parts of the liver [75,76]. Parallel CCR5 inhibition impairs the migration, activation and proliferation of activated hepatic stellate cells [76,77].
In a phase 2b trial on CVC in patients with NASH (NAS ≥4) with fibrosis (stages 1–3), patients (n=289) were randomly assigned CVC 150 mg or placebo. The primary outcome was a ≥2-point improvement in NAS and no worsening of fibrosis at 1 year (CENTAUR, NCT02217475) [27,78]. The primary endpoint of NAS improvement in the intention-to-treat population and resolution of NASH was achieved in a similar proportion of subjects on CVC (n=145) and placebo. However, the fibrosis endpoint was met in significantly more subjects on CVC than placebo (20% vs. 10%, P=0.02). Treatment benefits were greater in those with higher disease activity and fibrosis stage at baseline. After 1 year of CVC treatment, twice as many subjects achieved improvement in fibrosis and no worsening of NASH compared with placebo.
AURORA (NCT03028740), a randomized, double-blind, placebo-controlled, multicenter phase 3 study is ongoing to evaluate the efficacy and safety of CVC for the treatment of liver fibrosis in 2,000 NASH patients with stage 2/3 fibrosis (Table 1). The study will be conducted in two parts. Part 1 will examine the surrogate endpoint of improvement in fibrosis of at least one stage and no worsening of NASH at 12 months. Subjects from part 1 will continue into part 2 and additional subjects will be newly randomized in part 2 to determine long-term clinical outcomes—histopathologic progression to cirrhosis, liver-related clinical parameters, and all-cause mortality. The trial began in April 2017, and the results are expected in December 2024.
Thyroid hormone receptor-β agonist
1) Resmetirom (MGL-3196)
The THR-β is highly expressed in hepatocytes. NASH might be, in part, a condition of diminished liver thyroid hormone levels or hepatic hypothyroidism, and the incidence of clinical and subclinical hypothyroidism is higher in patients with NAFLD or NASH relative to age-matched controls [79,80]. THR-β stimulation is responsible for the beneficial metabolic effects on triglycerides and cholesterol levels as well as improvements in hepatic steatosis [79]. With a favorable cardiometabolic profile and the alleviation of hepatic steatosis, THR-β agonists are being investigated for the treatment of NASH.
Resmetirom (MGL-3196) is a liver-directed, orally active, selective THR-β agonist designed to improve NASH by increasing hepatic fat metabolism and reducing lipotoxicity. In a phase 2 study, patients with biopsy-confirmed NASH (fibrosis stages 1–3) and a hepatic fat fraction ≥10% by MRI-PDFF, were randomly assigned 2:1 to receive resmetirom 80 mg or matching placebo, orally once daily (NCT02912260) [28].
The primary outcome was the percentage of change from baseline in hepatic fat fraction assessed by MRI-PDFF at 12 weeks. Resmetirom treatment resulted in a significant reduction in the hepatic fat fraction after 12 and 36 weeks. Resmetirom was well tolerated but caused an increase in gastrointestinal adverse events, which were self-limited and did not result in study withdrawal.
A phase 3 trial involving 2,000 NASH patients with stage 2/3 fibrosis is ongoing (MAESTRO-NASH, NCT03900429) (Table 1). The primary outcome is the effect of resmetirom 80 or 100 mg compared to placebo on liver histology, and there is a composite long-term outcome of the number of patients with onset of any of the adjudicated events—cirrhosis, all-cause mortality, and liver-related clinical parameters. The trial began in March 2019, and the results are due in March 2024.
2) VK2809
VK2809 is a small-molecule prodrug of a potent THR-β agonist. VK2809 is selectively cleaved in hepatic tissue by cytochrome P450 isozyme 3A4, to release a pharmacologically active metabolite. In a phase 2a trial, patients with a liver fat content of ≥8% as assessed by MRI-PDFF, an LDL-C level of ≥110 mg/dL, and a triglyceride level of ≥120 mg/dL were randomized to receive oral VK2809 at 5 mg once daily, 10 mg once every other day, 10 mg once daily, or placebo for 12 weeks [81].
Patients receiving VK2809 experienced a significant reduction in liver fat content by MRI-PDFF, relative to placebo. The median relative change from baseline in liver fat content was 53.8% for VK2809 5 mg once daily (P=0.0001), 56.5% for VK2809 10 mg once every other day (P=0.0018), and 59.7% for VK2809 10 mg once daily (P=0.0004), versus 9.4% for placebo.
Based on these results, a phase 2b study is ongoing in 337 patients with biopsy proven NASH, to assess the efficacy and safety of VK2809 (1.0, 2.5, 5.0, 10 mg) versus placebo for 52 weeks. The primary outcome is the relative change in liver fat content (assessed by MRI-PDFF) from baseline to 12 weeks (VOYAGE, NCT04173065). The study began in November 2019.
Hepatic lipid metabolism
Stearoyl-CoA desaturase 1 inhibitor, aramchol
Arachidyl amido cholanoic acid (aramchol) is a novel synthetic lipid molecule that is a conjugate of cholic acid (a bile acid) and arachidic acid (a fatty acid). It inhibits SCD1, which converts saturated fatty acids into monounsaturated fatty acids (Fig. 1) [82]. Aramchol has been studied in a phase 2a trial over 3 months at doses of 100 and 300 mg daily. This study demonstrated a reduction in liver fat by magnetic resonance spectroscopy (MRS) after 3 months of treatment in the group receiving aramchol, 300 mg, compared with placebo, but not in the group receiving 100 mg. There was also a reduction in the ALT level and the compound was well tolerated [83]. Higher doses of aramchol (400 and 600 mg) were administrated to patients with biopsy proven NASH (n=247) without cirrhosis in a phase 2b trial that evaluated their effect on the hepatic triglyceride content using MRS and liver biopsy (ARREST, NCT02279524) [29]. There was a significant ≥5% reduction in liver fat content with 600 mg aramchol, 47%, compared with placebo, 24%. According to liver histology, NASH resolution without worsening of fibrosis occurred more often with aramchol, 600 mg, than placebo (16.7% vs. 5%; odds ratio, 4.74; 95% CI, 0.99 to 22.66). Although the study was underpowered for histological assessment, a dose-response trend toward NASH resolution and fibrosis improvement was seen. A phase 3/4, multinational, multicenter, double-blind, placebo-controlled study to evaluate the efficacy and safety of aramchol in subjects with NASH (fibrosis 2 or 3) who are overweight or obese and have prediabetes or T2DM (ARMOR, NCT04104321) has been proceeding since September 2019 (Table 1).
Liver X receptor-α inhibitor, oltipraz
Oltipraz, is a synthetic dithiolethione that functions as an antisteatotic agent by inhibiting liver X receptor α (LXRα) activity (Fig. 1) [84]. Dithiolethiones, a novel class of adenosine monophosphate-activated protein kinase (AMPK) activators, prevent insulin resistance by inhibiting AMPK-dependent p70 ribosomal S6 kinase-1 (S6K1). AMPK is a serine/threonine kinase that plays a crucial role in the regulation of carbohydrate and fat metabolism, and it may modulate LXRα activity and decrease the expression of sterol regulatory element binding protein 1c (SREBP-1c), a key regulator of lipid production [85]. Oltipraz possesses therapeutic potential for steatosis by activating AMPK and inactivating S6K1. It also expedites lipid oxidation by inhibiting LXRα activity and decreasing the expression of SREBP-1c in the liver [84]. In a current phase 2 study, patients with a liver fat content of >20% and hypertransaminasemia were randomized to three groups: placebo (n=22), 30 mg of oltipraz (n=22), or 60 mg of oltipraz (n=24) twice daily for 24 weeks (PMK-N01GI1, NCT01373554) [86]. The primary outcome was the change in the liver fat content assessed by MRS from baseline to 24 weeks.
Compared with the placebo group, oltipraz significantly reduced the liver fat content in a dose-dependent manner. However, the absolute changes in insulin resistance and the levels of liver enzymes, lipids, and cytokines were not significantly different among the groups. Also, the incidence of adverse events was comparable among the groups.
A multicenter, randomized, double-blind, placebo-controlled, parallel, phase 3 clinical trial is ongoing to evaluate the efficacy and safety of oltipraz in 144 patients with NAFLD (NCT04142749) (Table 1). The primary outcome is the variation in liver fat content by MRS at 24 weeks compared to baseline. The trial began in December 2019, and initial results are expected in October 2021.
Acetyl-coenzyme A carboxylase inhibitor, firsocostat (GS0976)
The regulation of de novo lipogenesis (DNL) plays a central role in fatty acid synthesis and catabolism. The rate-limiting step in DNL is conversion of acetyl-coenzyme A (acetyl-CoA) to malonyl-CoA by the enzyme acetyl-CoA carboxylase (ACC). ACC has two isoforms. The ACC1 isoform catalyzes the formation of malonyl-CoA, the main substrate for fatty acid biosynthesis in the cytosol. ACC2 is localized in mitochondria, where malonyl-CoA serves as a potent allosteric inhibitor of carnitine palmitoyl-transferase (CPT) 1, the carrier protein of fatty acids into mitochondria for β-oxidation [83,87]. Inhibition of ACC1 and ACC2 would be expected to reduce DNL and enhance mitochondrial β-oxidation, respectively, supporting ACC inhibition as a therapeutic target in NASH [88-90]. Firsocostat (GS-0976) is a liver-targeted, small-molecule allosteric inhibitor of both ACC1 and ACC2 in development for the treatment of NASH (Fig. 1).
In a recent phase 2 trial, 126 patients with hepatic steatosis of ≥8% based on MRI-PDFF and liver stiffness of ≥2.5 kPa, based on MRE, or historical biopsy consistent with NASH and F1–F3 fibrosis, were randomly assigned (2:2:1) to receive GS-0976 (20 mg), GS-0976 (5 mg), or placebo daily for 12 weeks. In that study, administration of 20 mg of GS-0976 was safe and led to a significant reduction in hepatic fat content by MRI-PDFF and a decreased serum level of tissue inhibitor of metalloproteinase 1 (TIMP1), a marker of fibrogenesis (NCT02856555) (Table 2) [91].
Anti-fibrotic agents
Galectin-3 antagonist, belapectin (GR-MD-02)
Galectins are carbohydrate-binding proteins belonging to the family of non-integrin β galactoside-binding lectins [92]. Galectin-3 (Gal-3) is the most prominent galectin secreted in disease, mainly by macrophages. Gal-3 via its intracellular (antiapoptotic, macrophage differentiation) and extracellular (chemokinetic/chemotactic factor) effects is relevant to the physiopathology of hepatic fibrosis caused by various chronic liver diseases [93-96]. Galectin inhibitors are a new class of agents that target both secreted and membrane-associated galectins by virtue of their high molecular weight [97]. Belapectin (GR-MD-02, galactoarabino-rhamnogalacturonate) is a complex carbohydrate molecule derived from a natural plant compound, which has oligosaccharide chains containing galactose residues and binds to galectin-3 and, to a lesser extent, galectin-1. A phase 1 study has shown that belapectin is safe and well tolerated at single and multiple doses of 2, 4, and 8 mg/kg in patients with well-characterized NASH and advanced fibrosis but not cirrhosis [98].
In a multicenter, randomized, double-blind, placebo-controlled phase 2b trial, 162 patients with NASH, cirrhosis, and portal hypertension (hepatic venous pressure gradient [HVPG] ≥6 mm Hg) were randomly assigned to receive biweekly infusions of belapectin 2 mg/kg (n=54), 8 mg/kg (n=54), or placebo (n=54) for 52 weeks (Table 2) [99]. The primary endpoint was the change in HVPG (−28) at the end of the 52 week period compared with baseline. Belapectin was safe but was not associated with a significant reduction in HVPG or fibrosis, compared with placebo. However, in a subgroup analysis of patients without esophageal varices, 2 mg/kg belapectin did reduce HVPG and the development of varices. A phase 3 study to evaluate the safety and efficacy of belapectin for the prevention of esophageal varices in patients with NASH cirrhosis without esophageal varices is being initiated (NCT 04365868).
COMBINATION THERAPY
NASH is a multifactorial disease involving different contributing mechanisms, with no approved therapies. As a future therapeutic direction, drug combinations are promising because of targeting multipul NASH pathways. Most drug combinations comprise metabolic, inflammatory, and fibrotic agents; alternatively, an antidiabetic may be included. Recently, there are various combination therapeutic options for NASH (Table 3).
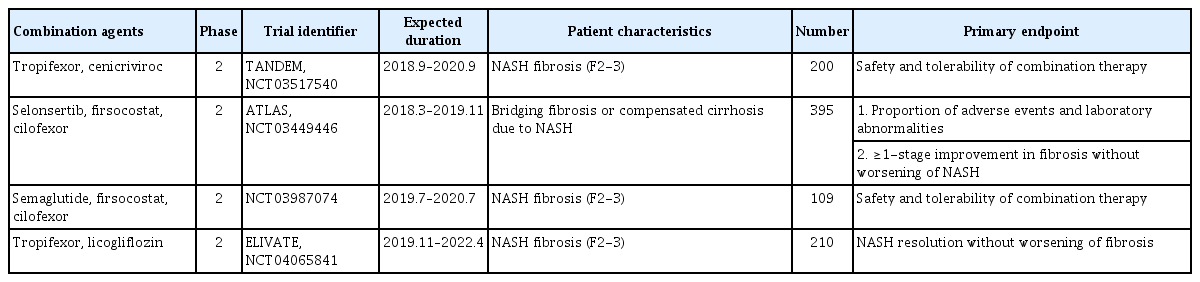
Selective combination treatments for NASH
TXR and CVC
A recent randomized, double-blind, multicenter, phase 2b study is ongoing to evaluate the safety and efficacy of the combination of TXR and CVC in 200 patients with NASH fibrosis (stages 2/3) (TANDEM, NCT03517540) (Table 3) [100]. Patients are randomized in a 1:1:1:1 ratio to receive TXR 140 μg once daily, CVC 150 mg once daily, TXR 140 μg+CVC 150 mg once daily, or TXR 90 μg+CVC 150 mg once daily. The study comprises a 48-week treatment period and a 4-week follow-up. The primary objective is evaluation of the safety and tolerability of the combination therapy compared with the monotherapies over 48 weeks. The secondary objective is efficacy, as defined by a ≥1-point improvement in liver fibrosis versus baseline or resolution of steatohepatitis after 48 weeks. This combination treatment targets the steatotic, inflammatory, and/or fibrotic pathways implicated in NASH.
Selonsertib, firsocostat, and cilofexor
A recent phase 2 study evaluated the safety and efficacy of a selonsertib, firsocostat, and cilofexor combination in patients with bridging fibrosis or compensated cirrhosis caused by NASH (ATLAS, NCT03449446) (Table 3). The primary objectives were to assess the safety and tolerability of selonsertib, firsocostat, and cilofexor, administered alone or in combination, and the changes in liver fibrosis without worsening of NASH. The data will be presented in 2020.
Semaglutide, firsocostat, and cilofexor
In a phase 2 study of the combination of semaglutide, firsocostat, and cilofexor, 109 patients with NASH fibrosis (stages 2/3) were randomly assigned to the semaglutide group, semaglu-tide+firsocostat group, semaglutide+cilofexor 30 mg group, semaglutide+cilofexor 100 mg group, or semaglutide+firsocostat+cilofexor group (NCT03987074) (Table 3). The primary objective was to evaluate the safety and tolerability of the study drug(s) in adult patients with NASH.
TXR and licogliflozin
Licogliflozin is a once-daily, oral, SGLT1/2 dual inhibitor. A phase 2, randomized, double-blind, parallel-group, multicenter study is assessing the efficacy, safety, and tolerability of oral TXR and licogliflozin combination therapy, compared to the monotherapies, in patients with NASH and liver fibrosis. The primary endpoint is the proportion of patients with resolution of NASH and no worsening of fibrosis or improvement in fibrosis by at least one stage without worsening of NASH at week 48 compared with baseline (ELIVATE, NCT04065841) (Table 3).
CONCLUSION
Although vitamin E, pioglitazone, and liraglutide improve liver histology, there is a no FDA approved drug for NASH. Therefore, weight loss by lifestyle modification, including diet and exercise, is the mainstay of NAFLD treatment. This review summarizes the pivotal phase 2 and 3 studies based on the mechanisms of action in NASH treatment (Fig. 2). Five pharmacologic agents—OCA, elafibranor, CVC, resmetirom, and aramchol—are under evaluation in large, histology-based phase 3 trials. Based on the results of these trials, new and effective drugs for NASH are expected within 2 to 4 years. Several phase 2 trials are ongoing for various agents, including non-bile acid FXR agonists, FGF19 and 21 analogues, GLP-1 RA, SGLT2 inhibitors, pan-PPAR agonists, MPC inhibitors, ACC inhibitors, and Gal-3 antagonists. Combination treatments are also being evaluated. Because NASH is a multifactorial disease, drug combinations show therapeutic potential. Finally, future treatment strategies will comprise combination treatments and precision medicine based on the different phenotypes of NASH and treatment response of the individual patient.
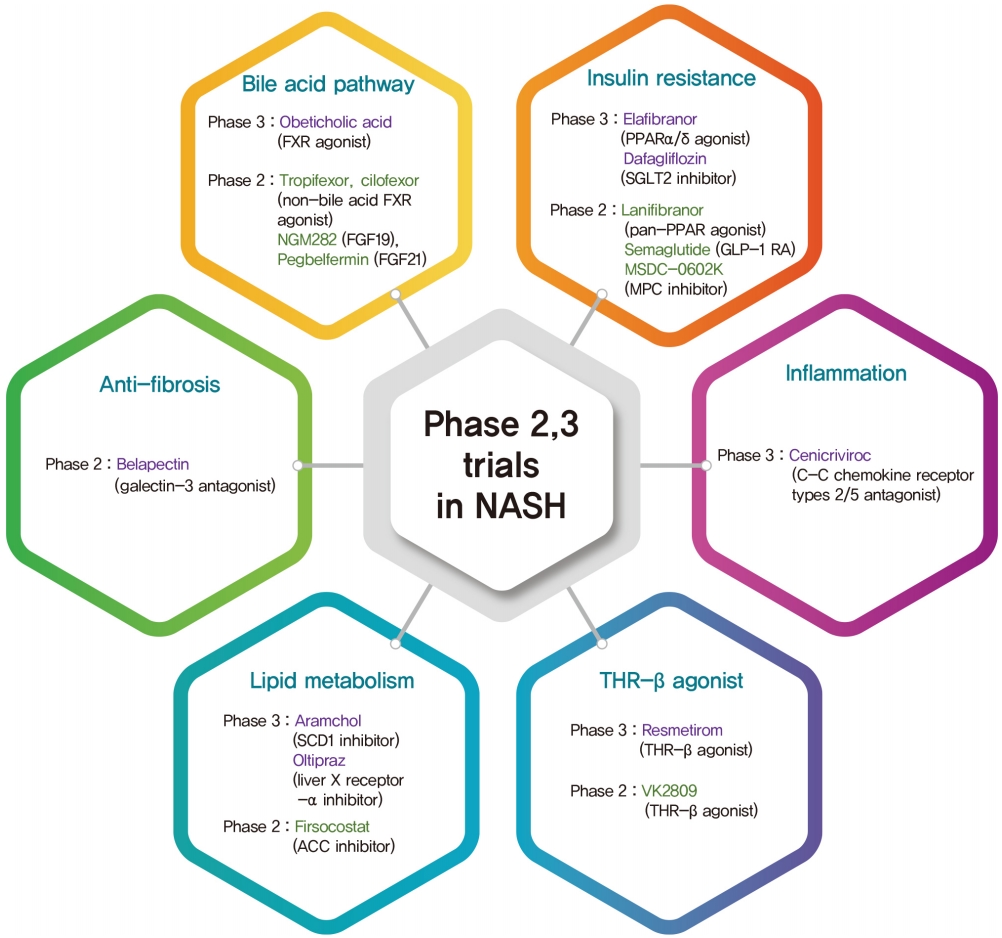
The classification of phase 2,3 trials based on mechanism of action in nonalcoholic steatohepatitis (NASH) treatment. FXR, farnesoid X receptor; FGF, fibroblast growth factor; PPAR, peroxisome proliferator-activated receptor; SGLT2, sodium glucose cotransporter 2; GLP-1 RA, glucagon-like peptide-1 receptor agonist; MPC, mitochondrial pyruvate carrier; SCD1, stearoylCoA desaturase 1; ACC, acetyl-coenzyme A carboxylase; THR-β, thyroid hormone receptor-β.
Notes
CONFLICTS OF INTEREST
No potential conflict of interest relevant to this article was reported.
Acknowledgements
This work was supported by the Soonchunhyang University Research Fund and the National Research Foundation of Korea grant funded by the Korea government (2020R1F1A1076282).