Adenine Nucleotide Translocator as a Regulator of Mitochondrial Function: Implication in the Pathogenesis of Metabolic Syndrome
Article information
Abstract
Mitochondria play key roles in energy production and intracellular reactive oxygen species (ROS) generation. Lines of evidence have shown that mitochondrial dysfunction contributes to the development of metabolic syndrome. The causes of mitochondrial dysfunction are complex, but overnutrition and sedentary living are among the best known causes of mitochondrial dysfunction. ATP synthesized in the mitochondria is exchanged for cytosolic ADP by adenine nucleotide translocator (ANT) to provide a continuous supply of ADP to mitochondria. We recently found that ANT function is essential for peroxisome proliferator-activated receptor-γ coactivator 1-α (PGC-1α)'s action on endothelial cells. PGC-1α is a transcriptional coactivator of nuclear receptors, playing an important role in fatty acid oxidation and mitochondrial biogenesis. Recent studies have shown that PGC-1α decreases intracellular ROS generation by increasing the expression of antioxidant genes. In our study, PGC-1α reduced cell apoptosis and ROS generation in endothelial cells by increasing ATP/ADP translocase activity of ANT and ANT1 expression. Here we review the role of ANT in maintaining proper mitochondrial function, and possible role of ANT dysfunction in the pathogenesis of metabolic syndrome.
INTRODUCTION
Metabolic syndrome is a cluster of common metabolic risk factors for atherosclerosis and type 2 diabetes occurring in obese subjects [1]. Metabolic syndrome is rapidly increasing in prevalence worldwide as a consequence of the continued obesity "epidemic", and as a result will have a considerable impact on the global incidence of cardiovascular disease and type 2 diabetes [2]. The major pathophysiologic mechanism of metabolic syndrome is insulin resistance [3]. Growing body of evidence has shown that mitochondrial dysfunction is an important pathogenic mechanism of diseases associated with insulin resistance, i.e., diabetes, atherosclerosis, and fatty liver disease [4].
Mitochondrion is the main organ of energy production, mostly in the form of ATP via oxidative phosphorylation (OXPHOS) and also a major site of intracellular reactive oxygen species (ROS) generation [5]. The ATP synthesized in the mitochondria is exchanged for cytosolic ADP by adenine nucleotide translocator (ANT) to provide a continuous supply of ADP to mitochondria. ATP/ADP exchange by ANT is essential for the maintenance of ATP synthase activity [6]. On the other hand, in states of impaired function of ATP/ADP exchange, ANT plays a major role in generating ROS and inducing cell apoptosis [7,8]. In this article, we will review the role of ANT in maintaining mitochondrial function, and possible role of ANT dysfunction in the pathogenesis of metabolic syndrome.
STRUCTURE AND FUNCTION OF MITOCHONDRIA
Structure
Mitochondrion is an intracellular double-membrane organelle present in most of eukaryotic cells [9]. Mitochondria form a reticulum that is in continuous communication through dynamic fusion and fission events, moving actively to different regions of the cell through interactions with the cytoskeleton [10]. The mitochondrial reticulum is composed of an inner and outer membrane, between which lies the intermembranous space, and a matrix contained within the inner membrane. The surface of the inner membrane is folded into cristae, which gives mitochondrion its characteristic morphology (Fig. 1).
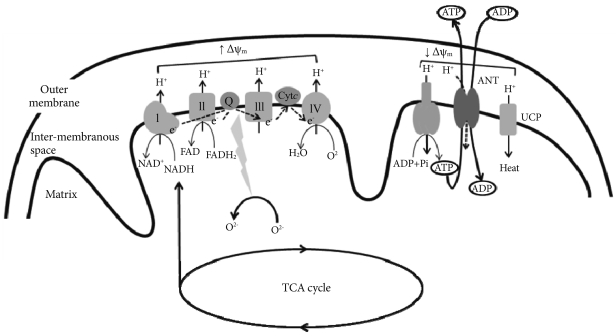
Mitochondrial electron-transport chain (ETC). Electrons derived from reducing equivalents (NADH and FADH2) are transported within ETC to molecular oxygen to produce water. As the electrons are transported, the free energy released is used to pump the protons into the intermembranous space. The proton gradient generated creates mitochondrial membrane potential (Δψm). The proton gradient produced is dissipated through the mitochondrial ATPase to produce ATP (OXPHOS or coupled respiration). The ATP synthesized in the mitochondria is exchanged for cytosolic ADP by adenine nucleotide translocator (ANT). Reactive oxygen species (ROS) is normally produced in the ETC during respiration, but delay of electron transport in the ETC results in the overproduction of ROS. ROS generation is more likely to occur when the proton gradient is large (increase in Δψm). Accumulation of ROS activates uncoupling protein (UCP), which dissipates the proton gradient without producing ATP (uncoupled respiration), decreases Δψm and ROS production. ANT also exhibits uncoupling activity or proton leak, and decreases ROS production and Δψm.
Mitochondrion has its own circular mitochondrial DNA (mtDNA) molecule, which encodes for 37 genes (13 of which are subunits of the electron transport chain, ETC) [11]. The majority of proteins regulating mitochondrial structure, function and biogenesis are encoded by the nuclear DNA. However, mtDNA gene products are essential for normal functioning of the mitochondrial respiratory chain. The mtDNA has no introns and is poorly equipped with repair mechanism, rendering it susceptible to oxidative damage and mutations. The mtDNA mutations accumulate with age, and these mutations might have an important role in the process of senescence and diabetes [12].
Function
Inner membrane contains all the transmembrane proteins of the ETC as well as the mitochondrial ATPase. The matrix contains the enzymatic machinery for TCA cycle, which provides reducing equivalents, such as NADH and FADH2, to the ETC, and for β-oxidation, which generates acetyl-CoA from acyl chains. The ETC is composed of four large multisubunit complexes (complex I to IV) with more than 85 individual gene products. Electrons derived from reducing equivalents NADH and FADH2 are transported within the ETC to molecular oxygen to produce water. As the electrons are transported, the free energy released is used to pump the protons into the inter-membranous space. The proton gradient thus generated creates electrochemical gradient across the inner mitochondrial membrane (mitochondrial membrane potential, Δψm) (Fig. 1). The energy contained in the proton gradient is then coupled to ATP production as protons flow back into the matrix through the mitochondrial ATPase. This process is known as OXPHOS, or coupled respiration [13]. The high proton gradient also drives the rapid entry of Ca2+ into the mitochondrial matrix, buffering its concentration in the cytoplasm [14].
Mitochondria are also a potent source of ROS [15]. ROS is normally produced in the ETC during respiration, but delay of electron transport in the ETC results in the overproduction of ROS. ROS generation is more likely to occur when the proton gradient is large: increase in Δψm is associated with delay of electron transport in ETC [16]. Consistent with this idea, we recently reported that overexpression of uncoupling protein 2 (UCP2), which dissipates the proton gradient without producing ATP, decreased Δψm and ROS production in cultured endothelial cells [17].
Mitochondria are also the prime regulator of apoptosis [12]. When confronted with cellular stress, mitochondria open the mitochondrial permeability transition pore (mtPTP) [18]. Opening of the mtPTP allows the release of mitochondrial proteins, such as cytochrome c, caspases, and apoptosis initiating factor, to induce apoptosis [19].
MITOCHONDRIAL DYSFUNCTION IN METABOLIC SYNDROME
Skeletal muscle is the major organ that determines whole body insulin sensitivity. In 1963, Randle et al. [20] proposed that increased free fatty acids (FFA) availability and oxidation lead to insulin resistance in skeletal muscle by inhibiting glucose oxidation and glycogen synthesis. Subsequent studies have shown that both glucose oxidation and glycogen synthesis are impaired in state of high FFA availability [21-23]. However, recent studies have suggested that defective intracellular fatty acid metabolism in skeletal muscle, rather than a simple oversupply of fatty acid fuel, is causally related to the development of insulin resistance [24]. In accordance with this concept, we previously showed that lipolysis in skeletal muscle was decreased in high fat-fed rats, suggesting that intracellular triglyceride accumulation in the insulin resistant state is the consequence of a diminished fatty acid oxidation capacity rather than the cause of insulin resistance [25].
Insulin resistance in the elderly or diabetic offspring is related to a reduction in the mitochondrial oxidative phosphorylation capacity [26,27]. Petersen et al. [26] reported that insulin resistance in the skeletal muscle of insulin resistant offspring of patients with type 2 diabetes was associated with dysregulation of intramyocellular fatty acid metabolism, possibly because of an inherited defect in mitochondrial oxidative phosphorylation. It was suggested that insulin resistance in humans arises from defects in mitochondrial fatty acid oxidation, which leads to increases in intracellular fatty acid metabolites (fatty acyl CoA and diacylglycerol) that disrupt insulin signaling [28].
OVERNUTRITION AS A CAUSE OF MITOCHONDRIAL DYSFUNCTION IN METABOLIC SYNDROME
The cause of mitochondrial dysfunction in metabolic syndrome may be multifactorial [29]. Among them, overnutrition and underutilization of nutrition are shown to induce mitochondrial dysfunction [30]. Chronic aerobic exercise increases mitochondrial content in muscle, thereby increasing ATP generating capacity [31]. On the other hand, chronic disuse of muscle decrease mitochondrial content and oxidative capacity leading to impaired glucose utilization [32].
It is well established that fasting prolongs lifespan. SIRT (mammalian homologues of Sir2; silent information regulator 2), which was identified as a mediator of longevity, increases mitochondrial biogenesis and improves mitochondrial function [33]. Conversely, high-fat diet (HFD) has been shown to reduce mitochondrial function. Genes necessary for OXPHOS and mitochondrial biogenesis were downregulated in skeletal muscle of the mice given HFD [34]. It was also shown that HFD decreases the expression of oxidative genes in healthy human individuals [34]. As a consequence of mitochondrial dysfunction and impaired fatty acid oxidation, intracellular levels of lipid metabolites, i.e., long chain fatty acyl coenzyme A (LCAC), diacylglycerol, and ceramides, are increased in skeletal muscle, liver, heart and pancreas β-cells of obese subjects [28]. Excess intake of nutrients also increases ROS production in the mitochondria. ROS-induced peroxidation may in turn leads to damage of mitochondria and further deterioration in oxidative capacity [35].
ATP/ADP TRANSLOCASE (ANT)
Human ANT has four isoforms (ANT1, ANT2, ANT3, and ANT4), which display distinct tissue-specific expression patterns. ANT1 is predominantly expressed in the heart, skeletal muscle, and brain. ANT2 is predominantly expressed in the liver and in cells with increased proliferative activity. ANT3 is ubiquitously detected. ANT4 is expressed in the liver, testis, and undifferentiated embryonic stem cells [36]. Among them, we will primarily focus on ANT1.
FUNCTIONS OF ANT ATP/ADP TRANSLOCASE(ANT)
ATP/ADP translocase
ANT is a protein complex of two subunits that is located in the inner mitochondrial membrane and facilitates the exchange of mitochondrial ATP and cytosolic ADP [37]. ANT provides a continuous supply of ADP necessary to maintain the oxidative phosphorylation process. ATP/ADP exchange by ANT plays an essential role for the maintenance of ATP synthase activity and normal levels of Δψm. Impaired ATP/ADP translocase activity of ANT decreases intramitochondrial ADP and ATP synthesis, and increases Δψm [6].
Regulation of apoptosis
As described above, the prime function of ANT is to facilitate the ATP/ADP exchange across the inner mitochondrial membrane. However, in states where ATP/ADP translocase activity is impaired, ANT plays a major role in promoting apoptosis.
Mitochondrial membrane permeabilization (MMP) is a rate limiting step of apoptosis and is mediated by the mitochondrial permeability transition pore (mtPTP) [38]. mtPTP is a nonspecific pore, permeable to all molecules of less than 1.5 kDa and is formed by the voltage-dependent anion channel (VDAC), members of the pro- and anti apoptotic Bax/Bcl2 protein family, cyclophilin D, and the ANT [39]. Additional proteins that were proposed to be part of the mtPTP complex are hexokinase, creatine kinase, and peripheral benzodiazepine receptor [40]. mtPTP opening causes swelling of the mitochondrial matrix and outer membrane rupture. This is followed by release of cytochrome c and other proapoptotic proteins into cytosol [38].
ANT has been widely accepted as a component for the mtPTP complex, which was first proposed by Halestrap et al. in 1990 [41]. ANT has been proposed to interact with VDAC, which is located in the outer mitochondrial membrane, to form a large protein-permeable conduit [42]. However, Wallace et al. have shown that mitochondria from livers of ANT-knockout mice, in which the ANT has been genetically inactivated, still possess mtPTP activity [18]. This would imply that the ANT is not an essential component of the mtPTP. Despite these debates, ANT is still considered to play a major regulatory role in the genesis of mtPTP [42].
Mitochondrial uncoupler
Under physiological conditions, mitochondrial oxygen consumption is tightly coupled to ATP synthesis. The bulk of proton re-enter the matrix via the F0F1 ATPase, which uses the energy to regenerate ATP from ADP (coupling of OXPHOS). A small proportion of proton can bypass the F0F1 ATPase, so that mitochondrial oxygen consumption is not coupled to ATP synthesis (mitochondrial uncoupling) [43]. In the 1970s, a protein responsible for non-shivering thermogenesis was identified in the inner mitochondrial membrane of brown adipose tissue mitochondria, and was named as uncoupling protein (UCP1) [44]. More recently, four more UCP homologues have been identified (UCP2, UCP3, UCP4 and UCP5/BCMP1 [brain mitochondrial carrier protein 1]) [45]. These proteins mediate proton leak across the mitochondrial membrane and decrease Δψm [46]. Since ROS production increases with increasing Δψm, UCP-mediated uncoupling has been proposed to play a role in decreasing mitochondrial ROS production [17]. This may represent a mechanism by which mitochondria protect themselves from oxidative damage [47].
Several lines of evidence suggested that ANT is also a mitochondrial uncoupler and is responsible for basal uncoupling or proton leak [43]. In rodents, ANT1 and ANT2 were shown to mediate uncoupling by fatty acids and to lower mitochondrial membrane potential in heart and skeletal muscle [48]. It was also demonstrated that ANT1-deficient mice have a 50% decrease in proton conductance in skeletal muscle [49]. In the heart, 4-hydroxy-2-nonenal-induced proton leak could be inhibited by the ANT inhibitor carboxyatractyloside, but not by the UCP inhibitor GDP [50]. These results suggest that ANT may decrease mitochondrial ROS production by functioning as an uncoupler.
INCREASED INTRACELLULAR LCAC IN METABOLIC SYNDROME IMPAIRS ATP/ADP TRANSLOCASE ACTIVITY
Central obesity is associated with increased cytosolic triglyceride stores in non-adipose tissue such as muscles, liver and pancreatic β-cells [51-53]. Cytosolic triglyceride is a source of LCAC, the metabolically active form of fatty acids. LCAC may accumulate under pathological conditions with excess lipid supply, such as obesity, and conditions with a mitochondrial fatty acid β-oxidation defect [54]. LCAC was shown to inhibit the ATP/ADP translocase activity of the ANT by competitive displacement of the nucleotide from its binding site on the protein [55].
It has been thus hypothesized that increased concentrations of LCACs in the cell interfere with mitochondrial function through inhibition of the ANT. Inhibition of ATP/ADP translocase activity lowers cytosolic ATP and matrix ADP availability, and increases mitochondrial membrane potential (Δψm) [8]. These events promote the formation of ROS, resulting in impaired cellular functions and cell death. Inhibition of the mitochondrial ANT by LCACs has been thus proposed to contribute to mitochondrial dysfunction in metabolic syndrome [8,56].
PGC-1α PREVENTS ENDOTHELIAL APOPTOSIS BY INCREASING ATP/ADP TRANSLOCASE ACTIVITY
Peroxisome proliferator-activated receptor-γ coactivator 1-α (PGC-1α) is a transcriptional coactivator of nuclear receptors, playing an important role in energy metabolism [57]. PGC-1α increases mitochondrial biogenesis and fatty acid oxidation [58]. In addition, recent studies have reported that PGC-1α is a major regulator of intracellular ROS generation. It was suggested that PGC-1α increases the expression of antioxidant genes [59].
We recently found that PGC-1α prevents endothelial apoptosis by increasing ATP/ADP translocase activity of ANT [7]. It is well known that fatty acids, such as linoleic acid (LA), increase ROS generation and cell apoptosis in endothelial cells [17]. LA treatment in human aortic endothelial cells increased intracellular and mitochondrial ROS generation and apoptosis. PGC-1α overexpression prevented LA-induced increases in ROS generation and apoptosis (Table 1, Fig. 2).

Effect of linoleic acid (LA) and PGC1-α on various functions of endothelial cells
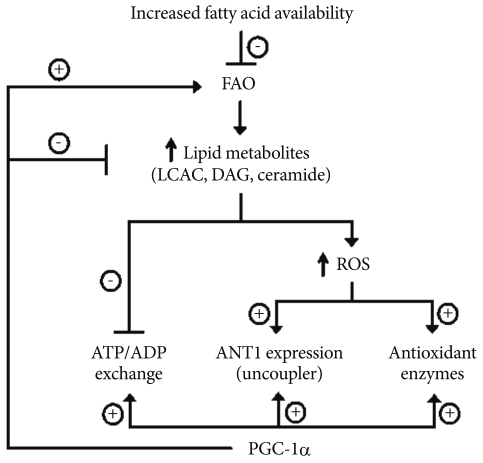
Proposed model of PGC-1α actions on endothelial cells to prevent ROS generation and cell apoptosis. PGC1-α, peroxisome proliferator-activated receptor-γ coactivator 1-α; ROS, reactive oxygen species; FAO, fatty acid oxidation; LCAC, long chain fatty acyl coenzyme A; DAG, diacylglycerol; ANT, adenine nucleotide translocator.
LA increased the mRNA and protein expression of antioxidant enzymes, including manganese superoxide dismutase, copper-zinc superoxide dismutase, catalase, glutathione peroxidase, and uncoupling protein2 UCP2, consistent with the notion that oxidative stress can induce cellular antioxidant responses [60]. As previously reported [59], adenoviral overexpression of PGC-1α also increased the mRNA and protein expression of the same antioxidant enzymes and UCP2. However, in the presence of LA, endogenous PGC-1α did not further increase the expression of antioxidant enzymes or UCP2. This result suggests that PGC-1α's effect on antioxidant genes may not fully explain its effect to decrease intracellular ROS production and cell apoptosis.
Similar to other antioxidant genes, LA significantly increased ANT-1 expression. On the other hand, LA significantly decreased ATP/ADP translocase activity, as measured by 14CADP import. This was associated with a significant increase in Δψm (hyperpolarization) and ROS generation. Interestingly, inhibitors of fatty acyl CoA synthase and ceramide synthase reduced LA-induced effects on ATP/ADP translocase activity, suggesting involvement of lipid metabolites, such as LCAC, diacylglycerol, and ceramide, in LA-induced impairment of ATP/ADP translocase activity. As expected, this was associated with changes in intracellular ceramides levels. On the other hand, antioxidant N-aceylcysteine prevented LA-induced ANT-1 expression, but did not affect ATP/ADP translocase activity regardless of LA treatment. These results suggest that increased ROS generation with LA may be responsible for the increase in ANT-1 expression. More importantly, changes in ANT-1 expression may not account for the decrease in ATP/ADP translocase activity with LA.
PGC-1α overexpression completely reversed LA-dependent decreases in ATP/ADP translocase activity, and prevented LA-induced changes in Δψm. PGC-1α also increased ANT-1 expression but did not increase ANT-1 expression above the levels induced by LA. In isolated aortic ring, LA treatment significantly decreased endothelium-dependent vascular relaxation. PGC-1α significantly inhibited LA-induced decreases in endothelium-dependent vasorelaxation, confirming that PGC-1α has antiatherogenic effects in vascular endothelial cells. Taken together, PGC-1α-dependent enhancement of ATP/ADP translocase activity of ANT is critically required for the beneficial effects of PGC-1α on endothelial function.
CONCLUSION
From this brief review, we have shown that ANT function is important in the maintenance of mitochondrial function. The prime function of ANT is exchange of ATP and ADP across the inner mitochondrial membrane, which is important for both ATP production and maintenance of normal Δψm. ANT also plays a role as an uncoupler. These two functions are important to protect the mitochondria from increased ROS generation associated with increased Δψm. However, in states where ATP/ADP translocase activity is impaired, ANT participates to play a role in the genesis of mtPTP and cell apoptosis. We have shown recently that PGC1-α regulates ROS generation and apoptosis in endothelial cells by enhancing ATP/ADP translocase activity of ANT. Understanding these mechanisms may help to find measures to prevent or treat metabolic syndrome.