- Current
- Browse
- Collections
-
For contributors
- For Authors
- Instructions to authors
- Article processing charge
- e-submission
- For Reviewers
- Instructions for reviewers
- How to become a reviewer
- Best reviewers
- For Readers
- Readership
- Subscription
- Permission guidelines
- About
- Editorial policy
Articles
- Page Path
- HOME > Diabetes Metab J > Volume 44(2); 2020 > Article
-
ReviewBasic Research Histone Deacetylase 9: Its Role in the Pathogenesis of Diabetes and Other Chronic Diseases
-
Siqi Hu1
 , Eun-Hee Cho2, Ji-Young Lee1
, Eun-Hee Cho2, Ji-Young Lee1 -
Diabetes & Metabolism Journal 2020;44(2):234-244.
DOI: https://doi.org/10.4093/dmj.2019.0243
Published online: March 24, 2020
1Department of Nutritional Sciences, University of Connecticut, Storrs, CT, USA.
2Department of Internal Medicine, Kangwon National University School of Medicine, Chuncheon, Korea.
- Corresponding author: Ji-Young Lee. Department of Nutritional Sciences, University of Connecticut, 27 Manter Road, Storrs, CT 06269, USA. ji-young.lee@uconn.edu
• Received: December 19, 2019 • Accepted: January 8, 2020
Copyright © 2020 Korean Diabetes Association
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- As a member of the class IIa histone deacetylases (HDACs), HDAC9 catalyzes the deacetylation of histones and transcription factors, commonly leading to the suppression of gene transcription. The activity of HDAC9 is regulated transcriptionally and post-translationally. HDAC9 is known to play an essential role in regulating myocyte and adipocyte differentiation and cardiac muscle development. Also, recent studies have suggested that HDAC9 is involved in the pathogenesis of chronic diseases, including cardiovascular diseases, osteoporosis, autoimmune disease, cancer, obesity, insulin resistance, and liver fibrosis. HDAC9 modulates the expression of genes related to the pathogenesis of chronic diseases by altering chromatin structure in their promotor region or reducing the transcriptional activity of their respective transcription factors. This review summarizes the current knowledge of the regulation of HDAC9 expression and activity. Also, the roles of HDAC9 in the pathogenesis of chronic diseases are discussed, along with potential underlying mechanisms.
- Histone deacetylases (HDACs) are the enzymes that catalyze the removal of acetyl groups from ε-N-acetyl-lysine on histone proteins H2A/B, H3, and H4. When acetyl groups are removed from histones, DNA molecules wrap up histone proteins more tightly, consequently inhibiting gene transcription [1]. Conventionally, histone proteins are known as unique HDAC substrates. However, studies have identified non-histone substrates of HDAC, which are involved in the regulation of cell cycle progression, cytoskeleton reorganization, RNA splicing, and nuclear transport [2].
- HDAC are categorized into five major classes based on their sequence homology to the yeast original enzymes [3]. Class I HDAC include HDAC1, HDAC2, HDAC3, and HDAC8; Class IIa HDAC are HDAC4, HDAC5, HDAC7, and HDAC9; and Class IIb HDAC are HDAC6 and HDAC10. Class III HDAC are also known as NAD+-dependent sirtuin 1–7. HDAC11 is the only class IV HDAC. Similar to other class IIa HDAC, HDAC9 shuttles in and out of the nucleus, which is a significant way to regulate its activity [4]. Also, while Class I, III, and IV HDAC are ubiquitously expressed, HDAC9 is abundant in the brain and skeletal muscles with lower expression in the liver, bone marrow, and spleen in humans [56].
- Six different transcriptional variants of human HDAC9 have been identified to date [567]. The full-length form of HDAC9 is composed of 23 coding exons, which can be translated into a polypeptide containing 1,011 amino acids [5]. Zhou et al. [7] reported an uncharacterized human protein that shares a 50% identity with the N-terminal amino acid sequence of HDAC4, which was designated as HDAC-related protein (HDRP). HDRP, a truncated splice isoform of HDAC9 that lacks of the acetylase domain, is also called myocyte enhancer factor-2 (MEF2)-interacting transcription repressor (MITR). HDRP is recognized as a transcriptional repressor that shares 50% amino acid sequence identity to human HDAC4 and HDAC5 and a homolog of Xenopus MITR [7]. A year later, the same research group reported the sequence of full-length HDAC9 along with another alternative splicing product of HDAC9, i.e., HDAC9a [5]. Compared to the full-length form, HDAC9a is 132 amino acids shorter at the C terminus. Petrie et al. [6] cloned three more splicing variants of HDAC9 , i.e., HDAC9Δexon7, HDAC9Δexon12, and HDAC9Δexon15, in COS cells, a monkey fibroblast-like cell line.
- Phenotypic analyses in global Hdac9 knockout and Hdac9 transgenic mice suggest that HDAC9 is a regulator of myocyte [8] and adipocyte differentiation [9] and it plays a crucial role in the development of cardiac muscle [10]. Moreover, HDAC9 has been linked to the pathological processes of human diseases, such as cardiovascular disease (CVD), autoimmune disease, and breast cancer [11121314]. The present review summarizes the current understanding of the regulation and functions of HDAC9, and its roles in the development of chronic diseases.
INTRODUCTION
- Based on the role of HDAC in chromatin remodeling, HDAC9 is generally considered as a negative regulator of gene transcription. It was shown that the transcriptional activity of Gal4 reduces when Gal4 co-expresses with HDRP in HEK293T cells [7]. This result indicates that HDAC9 can suppress gene transcription. Interestingly, genome-wide mapping data suggest that HDAC are involved in gene activation in human breast epithelial cell lines and primary T-cells [1516]. It is possible that HDAC9 also contributes to transcription activation.
- Repressive effect of HDAC9 on transcription by the induction of histone deacetylation
- HDAC9 may induce histone deacetylation to inhibit gene transcription by functioning as a scaffold in a transcriptional repressor complex [17]. It is commonly believed that HDAC induce histone deacetylation in the promoter of target genes to suppress gene transcription, when they are recruited by transcription factors or incorporated into multiprotein transcriptional complexes. Class IIa HDAC contain a predicted deacetylase activity in their C-terminus [18]. However, in vitro assays suggest that HDAC4, 5, and 7 lack intrinsic enzymatic activities [1920]. The HDAC activity of human HDAC9 isolated from HEK293T cells was approximately 10% of human HDAC4 enzyme activity, indicating that HDAC9 has low intrinsic HDAC activity [5]. Class I, IIb and IV HDAC possess a conservative catalytic tyrosine residue in their catalytic domain [21]. However, this tyrosine residue is replaced by histidine in vertebrate class IIa HDAC, which results in the reduction of their deacetylase activity [21]. It has been shown that the enzyme activity of class IIa HDAC relies on the association of their C-terminus with a transcriptional repressor complex containing HDAC3, silencing mediator for retinoid and thyroid receptors/nuclear receptor corepressor, and nuclear receptor co-repressor [1920]. Therefore, HDAC9 may be a pseudoenzyme and function as a scaffold for the recruitment of HDAC3 to a transcriptional repressor complex as do other class IIa HDAC [1922].
- All members of class II HDAC, including HDAC9, contain binding sites for MEF2 family in the N-terminus [23]. MEF2 family is transcriptional factors essential for myogenic differentiation [24]. According to Mejat et al. [25], MITR physically interacts with HDAC1 and HDAC3, attenuating denervation-induced histone H3 hyperacetylation and reduces the transcriptional activity of MEF2 in murine skeletal muscle. Skeletal muscle from Hdac9-deficient mice showed increased histone H3 acetylation on the promotor of MEF2 target genes compared to that of wild-type (WT) mice [25]. Also, overexpression of WT Mitr reduced MEF2 transcriptional activity while overexpression of mutant Mitr lacking in the MEF2 interaction domain abolished the reduction in mouse skeletal muscle [25]. This study demonstrates that HDAC9 is likely to suppress MEF2 transcriptional activity by inducing histone H3 deacetylation on the promoter of its target genes via its interaction with MEF2.
- C-terminal-binding protein (CtBP), a widely-expressed transcriptional co-repressor, is shown to be involved in the repressive effect of HDAC9 on the transcriptional activity of MEF2. MITR physically interacts with CtBP in a cell-free system as well as in HEK293T cells [26]. Compared to WT Mitr, transfection of mutant Mitr without CtBP binding activity had less suppression on MEF2C transcriptional activity by approximately 50% in 10T1/2 cells, a mouse fibroblast cell line, indicating that its binding to CtBP is required to inhibit the transcriptional activity of MEF2 [26]. Interestingly, the absence of CtBP binding activity did not inhibit MITR binding to other HDAC, including HDAC1, HDAC3, HDAC4, and HDAC5, in HEK293T cells. Therefore, MITR may repress MEF2 transcriptional activity by binding to other class I and IIa HDAC independent of CtBP.
- Deacetylation of transcription factors by HDAC9 for the suppression of the transcriptional activity
- Studies have shown that HDAC9 deacetylates transcriptional factors, resulting in the reduction of their transcriptional activity. Wong et al. [27] identified upstream stimulatory factor 1 (USF1), a transcription factor, as the first non-histone substrate of HDAC9. USF1 is essential for the transcriptional activation of lipogenic genes, such as fatty acid synthase (FASN) and mitochondrial glycerol-3-phosphate acyltransferase (GPAM), as sterol regulatory element-binding proteins-1c cannot bind its response elements without being recruited by USF1 [28]. In the absence of insulin, HDAC9 directly binds to USF1, which is constitutively bound to the promoter region of FASN and GPAM in cell-free conditions, HEK293F cells, HepG2 cells (a human hepatocellular carcinoma [HCC] cell line), and fasted mouse livers [27]. Transfection of HDAC9 in HEK293F cells inhibited acetylation in Lys237 of USF1, reducing the transcriptional activity of USF1. Signal transducer and activator of transcription 5 (STAT5), a transcription factor mediating interleukin 2 signaling [29], is another non-histone HDAC9 substrate. The acetylation and transcriptional activity of STAT5 increased in regulatory T-cells (Treg) from Hdac9 knockout mice when comparing to those from WT mice, indicating that HDAC9 deacetylates STAT5 to inhibiting the formation of transcriptionally active STAT5 dimer [30].
- Deacetylation of transcriptional factors by HDAC9 may also facilitate their degradation. In addition to acetylation, a lysine ε-amino group is also a site for other post-translational modifications, such as ubiquitination and sumoylation, which can facilitate protein degradation [31]. Acetylation in the lysine ε-amino group can stabilize proteins by competing with ubiquitination, consequently inhibiting proteasome-mediated protein degradation [32]. NK3 homeobox 2 (Nkx3.2) is a transcription factor that inhibits chondrocyte hypertrophic maturation during cartilage development [33]. Choi et al. [34] suggest that HDAC9-mediated deacetylation induced sumoylation and subsequent ubiquitination of Nkx3.2, resulting in its degradation in ATDC5 cells, a mouse chondrocyte cell line. It is likely that HDAC9 functions as a scaffold to recruit HDAC1 to deacetylate Nkx3.2 although it physically associates with Nkx3.2.
- Potential role of HDAC9 in transcriptional activation
- While HDAC are commonly regarded as suppressors of gene transcription, a previous genome-wide mapping study suggests that HDAC can also play a role in transcriptional activation [16]. HDAC1, 2, 3, and 6 are enriched in the chromatin of active genes, but not silent genes, with positive correlation with gene transcription in human T helper cells [16]. As histone hyperacetylation in transcribed regions can lead to chromatin destabilization and cryptic initiation of transcription, HDAC are required to keep an appropriate level of histone acetylation to poise genes for future activation [16]. Also, some transcription factors, such as β-catenin and CCAAT/enhancer-binding protein β, are activated by deacetylation [3536]. Our RNA-Seq analysis showed that Hdac9 deletion in primary mouse hepatic stellate cells (HSC) suppressed several genes while upregulating considerable numbers of genes (unpublished data). One possible explanation is that HDAC9 may inhibit the expression of transcriptional repressors.
ROLE OF HDAC9 IN GENE TRANSCRIPTION
- The activity of HDAC9 is tightly regulated transcriptionally, post-transcriptionally, and post-translationally as described below.
- Transcriptional regulation of HDAC9 expression
- While transcriptional regulation is an essential means to alter the activity of HDAC9, limited studies have been conducted to determine transcription factors that can control the transcription of HDAC9. During cardiac and skeletal muscle differentiation, Hdac9 transcription is up-regulated when myoblasts enter their differentiation pathway [37]. Members of the MEF2 family, i.e., MEF2A, MEF2C, and MEF2D, bind to the promoter of Hdac9 for the induction of its transcription [37]. Interestingly, although the MEF2 family is well-known transcription factors of all class IIa HDAC, they bind to different regions of class IIa HDAC genes. For instance, the MEF2 family bind to the enhancer regions of HDAC4 and HDAC7 while they bind to the promoter-proximal region of HDAC5 and HDAC9 [38]. Other transcription factors, including nuclear factor kappa-light-chain-enhancer of activated B-cells, early growth response protein 1, and paired-box containing 5, are suggested to induce HDAC9 expression [38]. However, future studies should be warranted to verify the role of the transcription factors mentioned above in the regulation of HDAC9 expression.
- Post-transcriptional regulation of HDAC9 expression
- In contrast to full-length HDAC9, its splicing variants do not contain post-translational modification sites, nuclear localization signal (NLS), or catalytic domain [567]. For instance, HDAC9Δexon7 lacks two serine residues in the NLS domain, resulting in its constitutive nuclear exclusion [6]. The removal of exon12 during splicing leads to the formation of HDAC9Δexon12, which cannot be sumoylated due to the lack of sumoylation sites. HDAC9 with partial deletion of exon 15, a possible product of RNA editing, loses a region within the deacetylase domain and the open reading frame of HDAC9 [6]. More studies are needed to determine the biological consequence of each splicing variants that may not be properly regulated at the post-transcriptional level.
- Regulation of HDAC9 activity by phosphorylation
- As a class IIa HDAC, HDAC9 generally exerts its function in the nucleus, and therefore its cytoplasmic retention may inhibit its activity [39]. Phosphorylation of HDAC9 is known to modulate its activity by altering the subcellular localization of HDAC9. For entry into the nucleus, the NLS of class IIa HDAC needs to bind to importin-α [38], which is an adaptor protein responsible for the nuclear import of proteins [40]. The phosphorylation of serine residues in the NLS sequence reduces its binding to importin-α, and thus inhibits the nuclear localization of class IIa HDAC [38]. Deng et al. [41] reported that minibrain-related kinase phosphorylates the NLS sequence in MITR at Ser243, leading to its cytoplasmic retention and, ultimately, the attenuation of MEF2 transcriptional suppression in C2C12 myoblasts. Ser253 in the NLS sequence of MITR has been identified as a substrate for protein kinase C-related kinase-1 [42]. While phosphorylation at this serine residue alone did not lead to MITR nuclear export, it was a priming step for the nuclear exclusion of MITR by calcium calmodulin-dependent protein kinase (CaMK) in COS cells [42].
- Moreover, phosphorylation of HDAC9 can facilitate its physical interaction with 14-3-3 chaperones [43]. Binding with 14-3-3 chaperones can mask the NLS, thereby preventing the nuclear translocation of HDAC9 [44]. In COS cells, CaMK phosphorylates MITR at Ser218 and Ser448, which facilitates 14-3-3 binding to disrupt the physical interaction between MITR and MEF2 [39]. Nevertheless, the transfection of constitutively activated CaMK changed the nuclear distribution of MITR, rather than inducing nuclear exclusion. These findings support that both phosphorylation in the NLS and 14-3-3 binding are required for the nuclear exclusion of MITR.
MECHANISMS FOR THE REGULATION OF HDAC9 ACTIVITY
- Zhang et al. [45] developed Hdac9 global knockout mice in 2002, which profoundly contributed to our understanding of HDAC9 functions in vivo. Mice with global Hdac9 deficiency showed no sign of cardiac abnormalities during early ages, but they developed spontaneous cardiac hypertrophy approximately 8 months after birth, indicating that the animals were sensitive to age-related hypertrophic signals [45]. Recent studies have also suggested the roles of HDAC9 in the development of various diseases, such as CVD, osteoporosis, autoimmune disease, cancer, obesity, and liver fibrosis, as described below.
- CVD
- Single nucleotide polymorphism (SNP) of HDAC9 is related to CVD risk in humans. Genome-wide association study (GWAS) for ischemic stroke in Europeans found that a SNP in HDAC9 (rs11984041) is positively associated with the risk of large vessel stroke [46]. Although later GWAS in the Chinese Han population did not find this SNP, another SNP of HDAC9, rs2389995, was found to be associated with decreased risk of ischemic stroke while rs2240419 was linked to the increased risk of ischemic stroke [47]. Also, compared with apolipoprotein E (ApoE) knockout mice, Hdac9 and ApoE double knockout mice exhibited remarkable decreases in aortic lesion sizes with less advanced lesions [48].
- Recent studies have been conducted to elucidate the underlying mechanisms by which HDAC9 contributes to increased susceptibility and progression of CVD. Cao et al. [49] demonstrated that Hdac9 deletion resulted in the attenuation of foam cell formation and atherosclerosis progression in low-density lipoprotein receptor (Ldlr) knockout mice. The atheroprotective effect of Hdac9 deletion occurred concomitantly with increased histone acetylation at the promoters of ATP-binding cassette transporter A1 (Abca1) and ATP binding cassette subfamily G member 1 (Abcg1) in thioglycollate-elicited mouse peritoneal macrophages, resulting in enhanced macrophage cholesterol efflux [49]. Furthermore, Han et al. [11] reported that HDAC9 knockdown inhibited oxidized-LDL-induced endothelial cell apoptosis in human umbilical vein endothelial cells (HUVECs), possibly decreasing the expression of pro-inflammatory cytokines, such as tumor necrosis factor-α and monocyte chemoattractant protein 1. Therefore, data available to date indicate that inhibition of HDAC9 may be a target to decrease CVD risk.
- Osteoporosis
- Evidence exists to support that HDAC9 inhibits osteoclast differentiation, which may inhibit bone resorption and thus protect against osteoporosis. Jin et al. [12] reported that Hdac9 knockout mice exhibited a lower bone mass and increased serum bone resorption markers compared to WT counterparts. Also, in vitro primary mouse osteoclast differentiation was facilitated by Hdac9 deletion, but diminished by Hdac9 overexpression [12]. This HDAC9 effect was linked to the transcriptional inhibition of peroxisome proliferator-activated receptor-gamma (Pparg), which is a critical regulator of osteoclast differentiation [1250].
- HDAC9 may protect against the development of age-related osteoporosis by promoting osteoblastogenesis of mesenchymal stem cells (MSC). According to Li et al. [51], a computational analysis predicts that microRNA-188 (miR-188) directly targets Hdac9 mRNA [49]. Overexpression of miR-188 suppresses Hdac9 translation in primary mouse bone marrow MSC (BMSC). In contrast, miR-188 deletion in mice reduced age-associated bone loss, higher osteoblastic bone formation, and lower bone marrow fat accumulation. Moreover, overexpression of miR-188 promoted differentiation of primary mouse BMSC into adipocytes (i.e., adipogenic differentiation) while inhibiting their differentiation into osteoblasts (i.e., osteogenic differentiation) [51]. The findings indicate that miR-188 may inhibit osteogenesis by silencing Hdac9. Also, Chen et al. [52] reported that overexpression of MITR favors osteogenesis but inhibits adipogenesis, while MITR knockdown leads to the opposing effects in 3A6 cells, a human MSC cell line, and primary human BMSC. Compared to being transfected alone, PPARγ transcriptional activity was reduced when it was co-transfected with MITR in 3A6 and HEK293 cells [52]. PPARγ facilitates MSC adipogenic differentiation while inhibits osteogenic differentiation; therefore MITR may promote MSC osteogenic differentiation by repressing PPARγ transcriptional activity [5253]. Hdac9 deficient mice appeared to have a decreased serum osteoblast number compared to their WT littermates, further supporting that HDAC9 facilitates osteoblastogenesis [12]. Overall, the evidence described above indicates that HDAC9 prevents osteoporosis by promoting osteogenesis while inhibiting osteoclastogenesis. However, data from human samples or clinical trials are needed to confirm these findings in humans.
- Autoimmune disease
- Previous reports suggest that HDAC9 is related to autoimmune diseases both in mice and humans. HDAC9 transcription was significantly increased in blood CD4+ T-cells from lupus patients compared with healthy controls [13]. Also, Hdac9 transcription was increased with lupus progression in total splenocytes and splenic CD4+ T-cell population from a murine disease model [13]. Furthermore, compared to control mice, colitis induced by dextran sodium sulfate was attenuated in mice with Hdac9 deficiency [54].
- The contribution of HDAC9 to autoimmune diseases was linked to its inhibitory effect on the immunosuppressive function of Treg. Forkhead box P3 (Foxp3) is a key regulator of the development and function of Treg [55]. Hdac9 deficiency in mice resulted in increased Foxp3+ expression in the colon and elevated the immunosuppressive function of Treg in vitro, consequently ameliorating colitis development [56]. Moreover, Hdac9 deficiency increased Treg proliferation and function by enhancing mitochondrial energy metabolism in mice via increased MEF2 binding to the promoter of PPARγ coactivator 1-alpha (PGC-1α), a principal regulator for mitochondrial biogenesis, with a concomitant increase in sirtuin 3 (SIRT3) protein [57]. PGC-1α and SIRT3 then increased oxidative phosphorylation in mouse Treg, which is essential for the proliferation and immunosuppressive function of Treg.
- Cancer
- Potential roles of HDAC9 in cancer progression have emerged recently. Huang et al. [14] demonstrated that Chinese female patients with high HDAC9 expression in their breast cancer tissues had reduced overall survival compared to those with a low HDAC9 level. In addition, HDAC9 expression was significantly upregulated in the breast cancer tissues collected from patients and human breast cancer cell lines (MCF-7, BT474). Importantly, HDAC9 knockdown using small interfering RNA (siRNA) significantly suppressed cell proliferation, migration, and invasion of MCF-7 and BT474 cells [14]. HDAC9 may promote breast cancer aggressiveness by suppressing the expression of miRNA that may contribute to the development of breast cancer. Breast cancer tissues with triple-negative breast cancer (TNBC), one of the most aggressive subtypes of breast cancer, have higher HDAC9 protein levels compared to non-TNBC cancer tissues [58]. Pan-class IIa HDAC inhibitor treatment or HDAC9 knockdown reduced the invasiveness while increasing miR-206 expression in human TNBC cell lines, e.g., MDA-MB-231, and HCC1143, and inhibited the angiogenesis of tumors in nude mice [58]. As miR-206 has been identified as a critical suppressor of TNBC [59], this study suggests that HDAC9 suppresses miR-206 expression in breast cancer cells and thereby promotes breast cancer cell invasion and breast cancer tissue angiogenesis.
- In addition to breast cancer cells, HDAC9 is known to suppress miRNA expression in other cancer cells. For instance, miR-376a was found to be down-regulated in HCC specimens [60]. Moreover, overexpression of miR-376a inhibited proliferation while inducing apoptosis in Huh7 cells, a human hepatocarcinoma cell line, indicating that miR-376a may inhibit the development of HCC [60]. HDAC9 knockdown in Huh7 cells remarkably increased miR-376a expression, suggesting that HDAC9 may repress the expression of miR-376a [61]. The knockdown also increased H3 histone acetylation at Lys18 in the differentially methylated region of maternally expressed 3 (Meg3-DMR), which is located upstream of the miR-376a gene, indicating HDAC9 may inhibit miR-376a expression by deacetylating Meg3-DMR [61].
- Obesity and insulin resistance
- Recent studies have established a link between HDAC9 and obesity/insulin resistance. Chatterjee et al. [9] demonstrated that HDAC9 functions as a negative regulator of adipogenic differentiation and Hdac9 expression is reduced in human and mouse pre-adipocytes before their adipogenic differentiation at both mRNA and protein levels. However, the reduction in Hdac9 expression before adipogenic differentiation was impaired in pre-adipocytes from high-fat diet-fed mice [62]. HDAC9 may mediate high-fat diet-induced inhibition of adipogenic differentiation. Hdac9 deletion protected mice from high-fat diet-induced weight gain, adipose tissue dysfunction, and liver steatosis while improving insulin sensitivity [62].
- Obesity-induced insulin resistance is known to lead to increases in gluconeogenesis via the activation of forkhead box O1 (FOXO1) [63], which is a transcription factor phosphoenolpyruvate carboxykinase (PEPCK) and glucose 6-phosphatase (G6PC) [64]. In chronic hepatitis C virus (HCV)-infected transgenic mice and patients, hepatic HDAC9 mRNA and protein levels were increased, and the patients had increased transcription and enzymatic activity of hepatic PEPCK [65]. HCV infection up-regulates the transcription of gluconeogenic enzymes, i.e., PEPCK and glucose G6PC, while HDAC9 knockdown diminished the induction in Huh7 cells [66]. Therefore, it seems that HDAC9 may mediate the HCV infection-induced gluconeogenic gene expression. Overexpression of HDAC9 leads to FOXO1 deacetylation and increases its transcriptional activity in Huh7 cells, while HDAC9 knockdown does the opposite [66]. These results suggest that HDAC9 increases FOXO1 transcriptional activity, possibly by deacetylation for the induction of gluconeogenic gene expression.
- Insulin treatment and refeeding increased the cytoplasmic retention of HDAC4 in primary mouse hepatocytes and HDAC4 phosphorylation in mouse livers [67]. Insulin signaling can activate salt-inducible kinase 2, which phosphorylates all class IIa HDAC for their cytoplasmic retention [68]. Therefore, it is possible that HDAC9 contributes to FOXO1-dependent upregulation of gluconeogenesis when insulin resistance occurs. Nevertheless, further experiments are required to test this hypothesis in animal models with diet-induced obesity and insulin resistance and human subjects with type 2 diabetes mellitus.
- Liver fibrosis
- HDAC9 may play essential roles in the activation of HSC and liver fibrosis. Activated HSC are major scar tissue-producing cells upon hepatic injury [69]. Our laboratory reported that HDAC9 mRNA and protein levels in primary mouse HSC increased during their in vitro activation [70]. Also, we demonstrated that HDAC9 mRNA and protein were up-regulated in human livers with primary biliary cirrhosis, nonalcoholic steatohepatitis, and alcoholic cirrhosis when compared to healthy livers [70]. Moreover, HDAC9 knockdown in LX-2 cells, a human HSC cell line, significantly decreased the expression of fibrogenic genes, such as alpha-actin 2 and collagen type I alpha one chain upon the stimulation by transforming growth factor β1, a potent fibrogenic cytokine. The studies further support that HDAC9 may be critical for HSC activation, and thus, inhibition of HDAC9 in HSC may prevent the development of liver fibrosis. An investigation is currently underway to test this hypothesis in our laboratory.
HDAC9 AND CHRONIC DISEASES
- By catalyzing the deacetylation of histones and transcription factors, HDAC9 plays a critical role in the regulation of gene expression (Fig. 1). While it is prevalent that HDAC9 silences gene expression, studies also exist to support that HDAC9 may be involved in transcriptional activation, although the underlying mechanism has been elusive. Mounting evidence suggests that HDAC9 is involved in the pathogenesis of CVD, osteoporosis, autoimmune disease, cancer, obesity, and liver fibrosis by altering the expression of genes critically involved in the pathogenic processes (Fig. 2). Therefore, HDAC9 may be a great molecular target for the prevention and therapy for the diseases. However, there are some challenges to overcome. Human HDAC9 gene encodes multiple protein isoforms and the role of each HDAC9 isoform in the development of chronic diseases has not been clearly understood. Also, the molecular mechanisms by which HDAC9 regulates the expression of mRNA and miRNA need further investigation.
CONCLUSIONS
-
Acknowledgements
- This study was supported by NIH 1R01DK108254-01 and USDA Multistate Hatch CONS00992 to Ji-Young Lee.
ACKNOWLEDGMENTS
-
CONFLICTS OF INTEREST: No potential conflict of interest relevant to this article was reported.
NOTES
- 1. Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res 2011;21:381-395. ArticlePubMedPMCPDF
- 2. Singh BN, Zhang G, Hwa YL, Li J, Dowdy SC, Jiang SW. Nonhistone protein acetylation as cancer therapy targets. Expert Rev Anticancer Ther 2010;10:935-954. ArticlePubMedPMC
- 3. Yang XJ, Seto E. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat Rev Mol Cell Biol 2008;9:206-218. ArticlePubMedPMCPDF
- 4. de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J 2003;370:737-749. ArticlePubMedPMCPDF
- 5. Zhou X, Marks PA, Rifkind RA, Richon VM. Cloning and characterization of a histone deacetylase, HDAC9. Proc Natl Acad Sci U S A 2001;98:10572-10577. ArticlePubMedPMC
- 6. Petrie K, Guidez F, Howell L, Healy L, Waxman S, Greaves M, Zelent A. The histone deacetylase 9 gene encodes multiple protein isoforms. J Biol Chem 2003;278:16059-16072. ArticlePubMed
- 7. Zhou X, Richon VM, Rifkind RA, Marks PA. Identification of a transcriptional repressor related to the noncatalytic domain of histone deacetylases 4 and 5. Proc Natl Acad Sci U S A 2000;97:1056-1061. ArticlePubMedPMC
- 8. Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet 2009;10:32-42. ArticlePubMedPMCPDF
- 9. Chatterjee TK, Idelman G, Blanco V, Blomkalns AL, Piegore MG Jr, Weintraub DS, Kumar S, Rajsheker S, Manka D, Rudich SM, Tang Y, Hui DY, Bassel-Duby R, Olson EN, Lingrel JB, Ho SM, Weintraub NL. Histone deacetylase 9 is a negative regulator of adipogenic differentiation. J Biol Chem 2011;286:27836-27847. ArticlePubMedPMC
- 10. Martin M, Kettmann R, Dequiedt F. Class IIa histone deacetylases: conducting development and differentiation. Int J Dev Biol 2009;53:291-301. ArticlePubMed
- 11. Han X, Han X, Wang Z, Shen J, Dong Q. HDAC9 regulates ox-LDL-induced endothelial cell apoptosis by participating in inflammatory reactions. Front Biosci (Landmark Ed) 2016;21:907-917. ArticlePubMed
- 12. Jin Z, Wei W, Huynh H, Wan Y. HDAC9 inhibits osteoclastogenesis via mutual suppression of PPARγ/RANKL signaling. Mol Endocrinol 2015;29:730-738. ArticlePubMedPMCPDF
- 13. Yan K, Cao Q, Reilly CM, Young NL, Garcia BA, Mishra N. Histone deacetylase 9 deficiency protects against effector T cell-mediated systemic autoimmunity. J Biol Chem 2011;286:28833-28843. ArticlePubMedPMC
- 14. Huang Y, Jian W, Zhao J, Wang G. Overexpression of HDAC9 is associated with poor prognosis and tumor progression of breast cancer in Chinese females. Onco Targets Ther 2018;11:2177-2184. PubMedPMC
- 15. Greer CB, Tanaka Y, Kim YJ, Xie P, Zhang MQ, Park IH, Kim TH. Histone deacetylases positively regulate transcription through the elongation machinery. Cell Rep 2015;13:1444-1455. ArticlePubMedPMC
- 16. Wang Z, Zang C, Cui K, Schones DE, Barski A, Peng W, Zhao K. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell 2009;138:1019-1031. ArticlePubMedPMC
- 17. Chatterjee TK, Basford JE, Yiew KH, Stepp DW, Hui DY, Weintraub NL. Role of histone deacetylase 9 in regulating adipogenic differentiation and high fat diet-induced metabolic disease. Adipocyte 2014;3:333-338. ArticlePubMedPMC
- 18. Lehmann LH, Worst BC, Stanmore DA, Backs J. Histone deacetylase signaling in cardioprotection. Cell Mol Life Sci 2014;71:1673-1690. ArticlePubMedPDF
- 19. Fischle W, Dequiedt F, Hendzel MJ, Guenther MG, Lazar MA, Voelter W, Verdin E. Enzymatic activity associated with class II HDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol Cell 2002;9:45-57. ArticlePubMed
- 20. Fischle W, Dequiedt F, Fillion M, Hendzel MJ, Voelter W, Verdin E. Human HDAC7 histone deacetylase activity is associated with HDAC3 in vivo. J Biol Chem 2001;276:35826-35835. ArticlePubMed
- 21. Lahm A, Paolini C, Pallaoro M, Nardi MC, Jones P, Neddermann P, Sambucini S, Bottomley MJ, Lo Surdo P, Carfi A, Koch U, De Francesco R, Steinkuhler C, Gallinari P. Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proc Natl Acad Sci U S A 2007;104:17335-17340. ArticlePubMedPMC
- 22. Sun Z, Feng D, Fang B, Mullican SE, You SH, Lim HW, Everett LJ, Nabel CS, Li Y, Selvakumaran V, Won KJ, Lazar MA. Deacetylase-independent function of HDAC3 in transcription and metabolism requires nuclear receptor corepressor. Mol Cell 2013;52:769-782. ArticlePubMed
- 23. Parra M. Class IIa HDACs: new insights into their functions in physiology and pathology. FEBS J 2015;282:1736-1744. PubMed
- 24. McKinsey TA, Zhang CL, Olson EN. Control of muscle development by dueling HATs and HDACs. Curr Opin Genet Dev 2001;11:497-504. ArticlePubMed
- 25. Mejat A, Ramond F, Bassel-Duby R, Khochbin S, Olson EN, Schaeffer L. Histone deacetylase 9 couples neuronal activity to muscle chromatin acetylation and gene expression. Nat Neurosci 2005;8:313-321. ArticlePubMedPDF
- 26. Zhang CL, McKinsey TA, Lu JR, Olson EN. Association of COOH-terminal-binding protein (CtBP) and MEF2-interacting transcription repressor (MITR) contributes to transcriptional repression of the MEF2 transcription factor. J Biol Chem 2001;276:35-39. ArticlePubMed
- 27. Wong RH, Chang I, Hudak CS, Hyun S, Kwan HY, Sul HS. A role of DNA-PK for the metabolic gene regulation in response to insulin. Cell 2009;136:1056-1072. ArticlePubMedPMC
- 28. Griffin MJ, Wong RH, Pandya N, Sul HS. Direct interaction between USF and SREBP-1c mediates synergistic activation of the fatty-acid synthase promoter. J Biol Chem 2007;282:5453-5467. ArticlePubMed
- 29. Mahmud SA, Manlove LS, Farrar MA. Interleukin-2 and STAT5 in regulatory T cell development and function. JAKSTAT 2013;2:e23154. ArticlePubMedPMC
- 30. Beier UH, Wang L, Han R, Akimova T, Liu Y, Hancock WW. Histone deacetylases 6 and 9 and sirtuin-1 control Foxp3+ regulatory T cell function through shared and isoform-specific mechanisms. Sci Signal 2012;5:ra45. ArticlePubMedPMC
- 31. Tan M, Luo H, Lee S, Jin F, Yang JS, Montellier E, Buchou T, Cheng Z, Rousseaux S, Rajagopal N, Lu Z, Ye Z, Zhu Q, Wysocka J, Ye Y, Khochbin S, Ren B, Zhao Y. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 2011;146:1016-1028. ArticlePubMedPMC
- 32. Caron C, Boyault C, Khochbin S. Regulatory cross-talk between lysine acetylation and ubiquitination: role in the control of protein stability. Bioessays 2005;27:408-415. ArticlePubMed
- 33. Caron MM, Emans PJ, Cremers A, Surtel DA, Coolsen MM, van Rhijn LW, Welting TJ. Hypertrophic differentiation during chondrogenic differentiation of progenitor cells is stimulated by BMP-2 but suppressed by BMP-7. Osteoarthritis Cartilage 2013;21:604-613. ArticlePubMed
- 34. Choi HJ, Kwon S, Kim DW. A post-translational modification cascade employing HDAC9-PIASy-RNF4 axis regulates chondrocyte hypertrophy by modulating Nkx3.2 protein stability. Cell Signal 2016;28:1336-1348. ArticlePubMed
- 35. Xu M, Nie L, Kim SH, Sun XH. STAT5-induced Id-1 transcription involves recruitment of HDAC1 and deacetylation of C/EBPbeta. EMBO J 2003;22:893-904. ArticlePubMedPMC
- 36. Wolf D, Rodova M, Miska EA, Calvet JP, Kouzarides T. Acetylation of beta-catenin by CREB-binding protein (CBP). J Biol Chem 2002;277:25562-25567. PubMed
- 37. Haberland M, Arnold MA, McAnally J, Phan D, Kim Y, Olson EN. Regulation of HDAC9 gene expression by MEF2 establishes a negative-feedback loop in the transcriptional circuitry of muscle differentiation. Mol Cell Biol 2007;27:518-525. ArticlePubMedPDF
- 38. Di Giorgio E, Brancolini C. Regulation of class IIa HDAC activities: it is not only matter of subcellular localization. Epigenomics 2016;8:251-269. ArticlePubMed
- 39. McKinsey TA, Zhang CL, Olson EN. Identification of a signal-responsive nuclear export sequence in class II histone deacetylases. Mol Cell Biol 2001;21:6312-6321. ArticlePubMedPMCPDF
- 40. Miyamoto Y, Yamada K, Yoneda Y. Importin α: a key molecule in nuclear transport and non-transport functions. J Biochem 2016;160:69-75. ArticlePubMed
- 41. Deng X, Ewton DZ, Mercer SE, Friedman E. Mirk/dyrk1B decreases the nuclear accumulation of class II histone deacetylases during skeletal muscle differentiation. J Biol Chem 2005;280:4894-4905. ArticlePubMed
- 42. Harrison BC, Huynh K, Lundgaard GL, Helmke SM, Perryman MB, McKinsey TA. Protein kinase C-related kinase targets nuclear localization signals in a subset of class IIa histone deacetylases. FEBS Lett 2010;584:1103-1110. ArticlePubMedPDF
- 43. Martin M, Kettmann R, Dequiedt F. Class IIa histone deacetylases: regulating the regulators. Oncogene 2007;26:5450-5467. ArticlePubMedPDF
- 44. Muslin AJ, Xing H. 14-3-3 Proteins: regulation of subcellular localization by molecular interference. Cell Signal 2000;12:703-709. ArticlePubMed
- 45. Zhang CL, McKinsey TA, Chang S, Antos CL, Hill JA, Olson EN. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell 2002;110:479-488. ArticlePubMedPMC
- 46. International Stroke Genetics Consortium (ISGC). Wellcome Trust Case Control Consortium 2 (WTCCC2). Bellenguez C, Bevan S, Gschwendtner A, Spencer CC, Burgess AI, Pirinen M, Jackson CA, Traylor M, Strange A, Su Z, Band G, Syme PD, Malik R, Pera J, Norrving B, Lemmens R, Freeman C, Schanz R, James T, Poole D, Murphy L, Segal H, Cortellini L, Cheng YC, Woo D, Nalls MA, Muller-Myhsok B, Meisinger C, Seedorf U, Ross-Adams H, Boonen S, Wloch-Kopec D, Valant V, Slark J, Furie K, Delavaran H, Langford C, Deloukas P, Edkins S, Hunt S, Gray E, Dronov S, Peltonen L, Gretarsdottir S, Thorleifsson G, Thorsteinsdottir U, Stefansson K, Boncoraglio GB, Parati EA, Attia J, Holliday E, Levi C, Franzosi MG, Goel A, Helgadottir A, Blackwell JM, Bramon E, Brown MA, Casas JP, Corvin A, Duncanson A, Jankowski J, Mathew CG, Palmer CN, Plomin R, Rautanen A, Sawcer SJ, Trembath RC, Viswanathan AC, Wood NW, Worrall BB, Kittner SJ, Mitchell BD, Kissela B, Meschia JF, Thijs V, Lindgren A, Macleod MJ, Slowik A, Walters M, Rosand J, Sharma P, Farrall M, Sudlow CL, Rothwell PM, Dichgans M, Donnelly P, Markus HS. Genome-wide association study identifies a variant in HDAC9 associated with large vessel ischemic stroke. Nat Genet 2012;44:328-333. ArticlePubMedPMCPDF
- 47. Qingxu G, Yan Z, Jiannan X, Yunlong L. Association between the gene polymorphisms of HDAC9 and the risk of atherosclerosis and ischemic stroke. Pathol Oncol Res 2016;22:103-107. ArticlePubMedPDF
- 48. Azghandi S, Prell C, van der Laan SW, Schneider M, Malik R, Berer K, Gerdes N, Pasterkamp G, Weber C, Haffner C, Dichgans M. Deficiency of the stroke relevant HDAC9 gene attenuates atherosclerosis in accord with allele-specific effects at 7p21.1. Stroke 2015;46:197-202. ArticlePubMed
- 49. Cao Q, Rong S, Repa JJ, St Clair R, Parks JS, Mishra N. Histone deacetylase 9 represses cholesterol efflux and alternatively activated macrophages in atherosclerosis development. Arterioscler Thromb Vasc Biol 2014;34:1871-1879. ArticlePubMedPMC
- 50. Wan Y, Chong LW, Evans RM. PPAR-gamma regulates osteoclastogenesis in mice. Nat Med 2007;13:1496-1503. ArticlePubMedPDF
- 51. Li CJ, Cheng P, Liang MK, Chen YS, Lu Q, Wang JY, Xia ZY, Zhou HD, Cao X, Xie H, Liao EY, Luo XH. MicroRNA-188 regulates age-related switch between osteoblast and adipocyte differentiation. J Clin Invest 2015;125:1509-1522. ArticlePubMedPMC
- 52. Chen YH, Yeh FL, Yeh SP, Ma HT, Hung SC, Hung MC, Li LY. Myocyte enhancer factor-2 interacting transcriptional repressor (MITR) is a switch that promotes osteogenesis and inhibits adipogenesis of mesenchymal stem cells by inactivating peroxisome proliferator-activated receptor gamma-2. J Biol Chem 2011;286:10671-10680. PubMedPMC
- 53. Sun H, Kim JK, Mortensen R, Mutyaba LP, Hankenson KD, Krebsbach PH. Osteoblast-targeted suppression of PPARγ increases osteogenesis through activation of mTOR signaling. Stem Cells 2013;31:2183-2192. ArticlePubMedPDF
- 54. Tao R, de Zoeten EF, Ozkaynak E, Chen C, Wang L, Porrett PM, Li B, Turka LA, Olson EN, Greene MI, Wells AD, Hancock WW. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat Med 2007;13:1299-1307. ArticlePubMedPDF
- 55. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol 2003;4:330-336. ArticlePubMedPDF
- 56. de Zoeten EF, Wang L, Sai H, Dillmann WH, Hancock WW. Inhibition of HDAC9 increases T regulatory cell function and prevents colitis in mice. Gastroenterology 2010;138:583-594. ArticlePubMed
- 57. Beier UH, Angelin A, Akimova T, Wang L, Liu Y, Xiao H, Koike MA, Hancock SA, Bhatti TR, Han R, Jiao J, Veasey SC, Sims CA, Baur JA, Wallace DC, Hancock WW. Essential role of mitochondrial energy metabolism in Foxp3+ T-regulatory cell function and allograft survival. FASEB J 2015;29:2315-2326. ArticlePubMedPMCPDF
- 58. Salgado E, Bian X, Feng A, Shim H, Liang Z. HDAC9 overexpression confers invasive and angiogenic potential to triple negative breast cancer cells via modulating microRNA-206. Biochem Biophys Res Commun 2018;503:1087-1091. ArticlePubMedPMC
- 59. Liang Z, Bian X, Shim H. Downregulation of microRNA-206 promotes invasion and angiogenesis of triple negative breast cancer. Biochem Biophys Res Commun 2016;477:461-466. ArticlePubMedPMC
- 60. Zheng Y, Yin L, Chen H, Yang S, Pan C, Lu S, Miao M, Jiao B. miR-376a suppresses proliferation and induces apoptosis in hepatocellular carcinoma. FEBS Lett 2012;586:2396-2403. ArticlePubMed
- 61. Zheng Y, Chen H, Yin M, Ye X, Chen G, Zhou X, Yin L, Zhang C, Ding B. MiR-376a and histone deacetylation 9 form a regulatory circuitry in hepatocellular carcinoma. Cell Physiol Biochem 2015;35:729-739. ArticlePubMedPDF
- 62. Chatterjee TK, Basford JE, Knoll E, Tong WS, Blanco V, Blomkalns AL, Rudich S, Lentsch AB, Hui DY, Weintraub NL. HDAC9 knockout mice are protected from adipose tissue dysfunction and systemic metabolic disease during high-fat feeding. Diabetes 2014;63:176-187. ArticlePubMedPDF
- 63. Gross DN, van den Heuvel AP, Birnbaum MJ. The role of FoxO in the regulation of metabolism. Oncogene 2008;27:2320-2336. ArticlePubMedPDF
- 64. Zou Y, Gong N, Cui Y, Wang X, Cui A, Chen Q, Jiao T, Dong X, Yang H, Zhang S, Fang F, Chang Y. Forkhead box P1 (FOXP1) transcription factor regulates hepatic glucose homeostasis. J Biol Chem 2015;290:30607-30615. ArticlePubMedPMC
- 65. Chen J, Wang N, Dong M, Guo M, Zhao Y, Zhuo Z, Zhang C, Chi X, Pan Y, Jiang J, Tang H, Niu J, Yang D, Li Z, Han X, Wang Q, Chen X. The metabolic regulator histone deacetylase 9 contributes to glucose homeostasis abnormality induced by hepatitis C virus infection. Diabetes 2015;64:4088-4098. ArticlePubMedPDF
- 66. Chen J, Zhang Z, Wang N, Guo M, Chi X, Pan Y, Jiang J, Niu J, Ksimu S, Li JZ, Chen X, Wang Q. Role of HDAC9-FoxO1 axis in the transcriptional program associated with hepatic gluconeogenesis. Sci Rep 2017;7:6102. ArticlePubMedPMCPDF
- 67. Mihaylova MM, Vasquez DS, Ravnskjaer K, Denechaud PD, Yu RT, Alvarez JG, Downes M, Evans RM, Montminy M, Shaw RJ. Class IIa histone deacetylases are hormone-activated regulators of FOXO and mammalian glucose homeostasis. Cell 2011;145:607-621. ArticlePubMedPMC
- 68. Walkinshaw DR, Weist R, Kim GW, You L, Xiao L, Nie J, Li CS, Zhao S, Xu M, Yang XJ. The tumor suppressor kinase LKB1 activates the downstream kinases SIK2 and SIK3 to stimulate nuclear export of class IIa histone deacetylases. J Biol Chem 2013;288:9345-9362. ArticlePubMedPMC
- 69. Moreira RK. Hepatic stellate cells and liver fibrosis. Arch Pathol Lab Med 2007;131:1728-1734. ArticlePubMedPDF
- 70. Yang Y, Bae M, Park YK, Lee Y, Pham TX, Rudraiah S, Manautou J, Koo SI, Lee JY. Histone deacetylase 9 plays a role in the antifibrogenic effect of astaxanthin in hepatic stellate cells. J Nutr Biochem 2017;40:172-177. ArticlePubMed
REFERENCES
Fig. 1
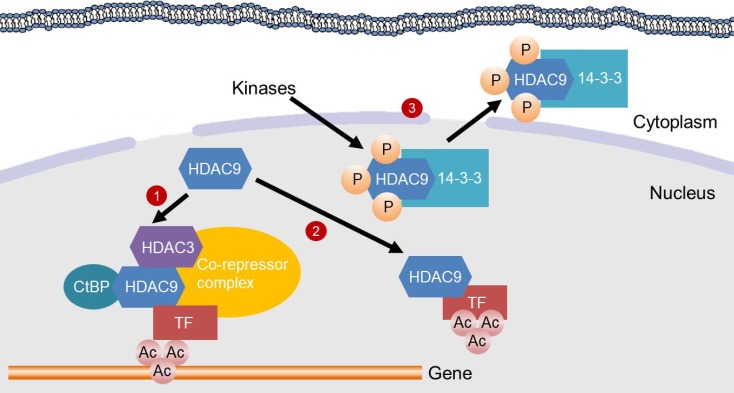
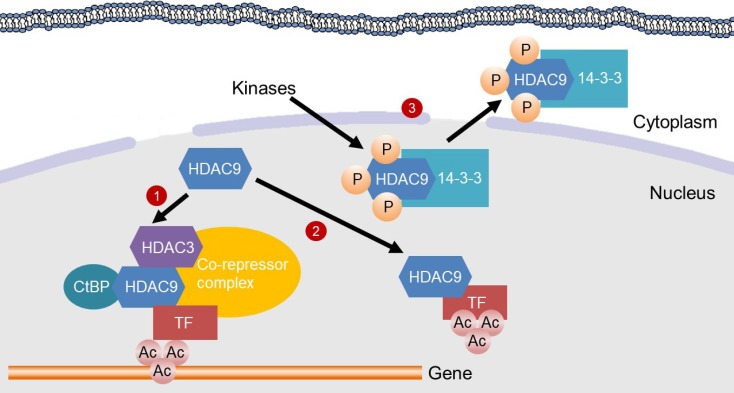
Mechanisms for the inhibition of gene transcription by histone deacetylase 9 (HDAC9). HDAC9 inhibits gene transcription by inducing deacetylation of transcription factors (TFs) or in the promoter region of genes. (1) HDAC9 directly binds to C-terminal-binding protein (CtBP) and HDAC3, which are recruited to a TF to assemble a co-repressor complex. Then, the co-repressor complex deacetylates histones in the promoter region of a gene, thereby suppressing gene transcription. (2) HDAC9 binds to a TF for its deacetylation, which inhibits the transcriptional activity. (3) Kinases, such as minibrain-related kinase and calcium calmodulin-dependent protein kinase, phosphorylate the nuclear localization signal region of HDAC9 for subsequent 14-3-3 binding, which leads to the exit of HDAC9 from the nucleus and therefore a loss of its activity.
Fig. 2
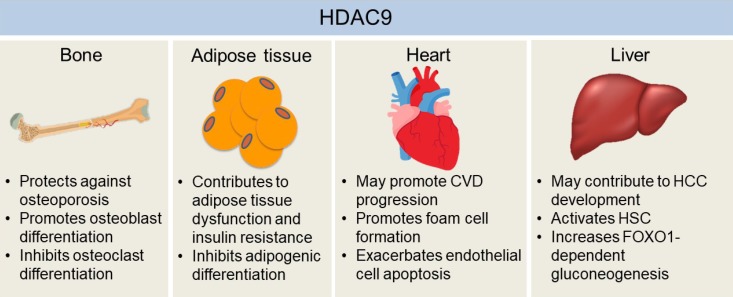
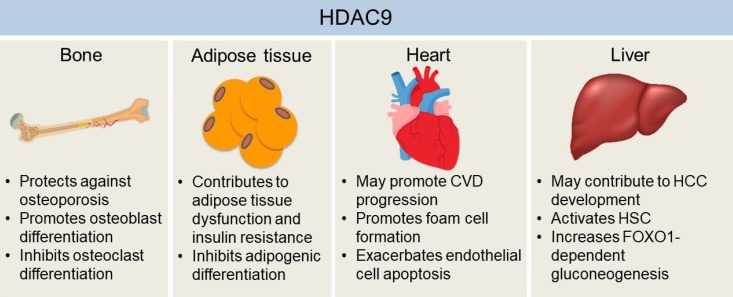
The effects of histone deacetylase 9 (HDAC9) on the development of chronic diseases in bone, adipose tissue, heart, and liver. HDAC9 facilitates osteoblast activity while inhibiting osteoclast differentiation, which may explain the protective effects of HDAC9 against osteoporosis in mice. Adipogenic differentiation of pre-adipocytes may help to alleviate hypoxia and dysfunctions of hypertrophied adipose tissue, which is inhibited by HDAC9. Also, mice with the deletion of Hdac9 are protected from diet-induced adipose tissue dysfunctions and systemic insulin resistance. Hdac9 deficiency also enhances cholesterol efflux from macrophages while inhibiting oxidized low-density lipoprotein-induced human endothelial cell apoptosis, indicating HDAC9 may exacerbate cardiovascular disease (CVD) development. Moreover, HDAC9 may contribute to hepatocellular carcinoma (HCC) development by suppressing miR-376a expression, a microRNA that induces the apoptosis of hepatocellular carcinoma cells. HDAC9 is also critical for hepatic stellate cell (HSC) activation and forkhead box O 1 (FOXO1)-dependent gluconeogenesis in the liver. Therefore, it may also be involved in the development of fibrosis and obesity-induced insulin resistance in the liver.
Figure & Data
References
Citations
Citations to this article as recorded by 
- Impact of housing temperature on adipose tissue HDAC9 expression and adipogenic differentiation in high fat‐fed mice
Samah Ahmadieh, Brandee Goo, Abdalrahman Zarzour, David Kim, Hong Shi, Praneet Veerapaneni, Ronnie Chouhaita, Nicole K. H. Yiew, Carla Dominguez Gonzalez, Akash Chakravartty, James Pennoyer, Nazeera Hassan, Tyler W. Benson, Mourad Ogbi, David J. Fulton, R
Obesity.2024; 32(1): 107. CrossRef - HDAC9 inhibition reduces skeletal muscle atrophy and enhances regeneration in mice with cigarette smoke-induced COPD
Guixian Zheng, Chao Li, Xiaoli Chen, Zhaohui Deng, Ting Xie, Zengyu Huo, Xinyan Wei, Yanbing Huang, Xia Zeng, Yu Luo, Jing Bai
Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease.2024; 1870(3): 167023. CrossRef - Identification of HDAC9 and ARRDC4 as potential biomarkers and targets for treatment of type 2 diabetes
Jing Liu, Lingzhen Meng, Zhihong Liu, Ming Lu, Ruiying Wang
Scientific Reports.2024;[Epub] CrossRef - HDAC9 as a Privileged Target: Reviewing its Role in Different Diseases
and Structure-activity Relationships (SARs) of its Inhibitors
Totan Das, Samima Khatun, Tarun Jha, Shovanlal Gayen
Mini-Reviews in Medicinal Chemistry.2024; 24(7): 767. CrossRef - Targeting histone deacetylases for cancer therapy: Trends and challenges
Tao Liang, Fengli Wang, Reham M. Elhassan, Yongmei Cheng, Xiaolei Tang, Wengang Chen, Hao Fang, Xuben Hou
Acta Pharmaceutica Sinica B.2023; 13(6): 2425. CrossRef - Therapeutic approach of natural products that treat osteoporosis by targeting epigenetic modulation
Guokai Zhang, Zhenying Liu, Zihan Li, Bing Zhang, Pengyu Yao, Yun Qiao
Frontiers in Genetics.2023;[Epub] CrossRef - Research Progress on Histone Deacetylase Inhibitors
玉姜 汤
Hans Journal of Medicinal Chemistry.2023; 11(02): 116. CrossRef - HDAC9 Inhibition as a Novel Treatment for Stroke
Hugh S. Markus
Stroke.2023; 54(12): 3182. CrossRef - Histone deacetylase 9 exacerbates podocyte injury in hyperhomocysteinemia through epigenetic repression of Klotho
Min Liu, Yang Zhang, Ping Zhan, Wenjuan Sun, Chuanqiao Dong, Xiaohan Liu, Yujie Yang, Xiaojie Wang, Yusheng Xie, Chengjiang Gao, Huili Hu, Benkang Shi, Ziying Wang, Chun Guo, Fan Yi
Pharmacological Research.2023; 198: 107009. CrossRef - Molecular mechanism and therapeutic potential of HDAC9 in intervertebral disc degeneration
Ming Lei, Hui Lin, Deyao Shi, Pan Hong, Hui Song, Bomansaan Herman, Zhiwei Liao, Cao Yang
Cellular & Molecular Biology Letters.2023;[Epub] CrossRef - Interindividual variability in transgene mRNA and protein production following adeno-associated virus gene therapy for hemophilia A
Sylvia Fong, Bridget Yates, Choong-Ryoul Sihn, Aras N. Mattis, Nina Mitchell, Su Liu, Chris B. Russell, Benjamin Kim, Adebayo Lawal, Savita Rangarajan, Will Lester, Stuart Bunting, Glenn F. Pierce, K. John Pasi, Wing Yen Wong
Nature Medicine.2022; 28(4): 789. CrossRef - Active RhoA Exerts an Inhibitory Effect on the Homeostasis and Angiogenic Capacity of Human Endothelial Cells
Michael Hauke, Robert Eckenstaler, Anne Ripperger, Anna Ender, Heike Braun, Ralf A. Benndorf
Journal of the American Heart Association.2022;[Epub] CrossRef - HDAC9 Contributes to Serous Ovarian Cancer Progression through Regulating Epithelial–Mesenchymal Transition
Long Xu, Jian Wang, Buhan Liu, Jiaying Fu, Yuanxin Zhao, Sihang Yu, Luyan Shen, Xiaoyu Yan, Jing Su
Biomedicines.2022; 10(2): 374. CrossRef - Common protein-coding variants influence the racing phenotype in galloping racehorse breeds
Haige Han, Beatrice A. McGivney, Lucy Allen, Dongyi Bai, Leanne R. Corduff, Gantulga Davaakhuu, Jargalsaikhan Davaasambuu, Dulguun Dorjgotov, Thomas J. Hall, Andrew J. Hemmings, Amy R. Holtby, Tuyatsetseg Jambal, Badarch Jargalsaikhan, Uyasakh Jargalsaikh
Communications Biology.2022;[Epub] CrossRef - Proposed minimal essential co-expression and physical interaction networks involved in the development of cognition impairment in human mid and late life
Zahra Salehi, Masoud Arabfard, Omid Sadatpour, Mina Ohadi
Neurological Sciences.2021; 42(3): 951. CrossRef - Emerging roles of SIRT6 in human diseases and its modulators
Gang Liu, Haiying Chen, Hua Liu, Wenbo Zhang, Jia Zhou
Medicinal Research Reviews.2021; 41(2): 1089. CrossRef - Quis Custodiet Ipsos Custodes (Who Controls the Controllers)? Two Decades of Studies on HDAC9
Claudio Brancolini, Eros Di Giorgio, Luigi Formisano, Teresa Gagliano
Life.2021; 11(2): 90. CrossRef - circ_0003204 Regulates Cell Growth, Oxidative Stress, and Inflammation in ox-LDL-Induced Vascular Endothelial Cells via Regulating miR-942-5p/HDAC9 Axis
Huan Wan, Ting You, Wei Luo
Frontiers in Cardiovascular Medicine.2021;[Epub] CrossRef - Histone deacetylase (HDAC) 9: versatile biological functions and emerging roles in human cancer
Chun Yang, Stéphane Croteau, Pierre Hardy
Cellular Oncology.2021; 44(5): 997. CrossRef - Dual HDAC/BRD4 inhibitors against cancer
Negar Omidkhah, Farzin Hadizadeh, Razieh Ghodsi
Medicinal Chemistry Research.2021; 30(10): 1822. CrossRef - miR‐211‐5p is down‐regulated and a prognostic marker in bladder cancer
Weisheng Wang, Zhiming Liu, Xuegang Zhang, Junning Liu, Junqing Gui, Maorong Cui, Yong Li
The Journal of Gene Medicine.2020;[Epub] CrossRef
 PubReader
PubReader Cite
Cite