Fuel-Stimulated Insulin Secretion Depends upon Mitochondria Activation and the Integration of Mitochondrial and Cytosolic Substrate Cycles
Article information
Abstract
The pancreatic islet β-cell is uniquely specialized to couple its metabolism and rates of insulin secretion with the levels of circulating nutrient fuels, with the mitochondrial playing a central regulatory role in this process. In the β-cell, mitochondrial activation generates an integrated signal reflecting rates of oxidativephosphorylation, Kreb's cycle flux, and anaplerosis that ultimately determines the rate of insulin exocytosis. Mitochondrial activation can be regulated by proton leak and mediated by UCP2, and by alkalinization to utilize the pH gradient to drive substrate and ion transport. Converging lines of evidence support the hypothesis that substrate cycles driven by rates of Kreb's cycle flux and by anaplerosis play an integral role in coupling responsive changes in mitochondrial metabolism with insulin secretion. The components and mechanisms that account for the integrated signal of ATP production, substrate cycling, the regulation of cellular redox state, and the production of other secondary signaling intermediates are operative in both rodent and human islet β-cells.
INTRODUCTION
Mitochondria are key to the unique and specialized ability of the pancreatic islet β-cell to couple metabolism and rates of insulin secretion with the levels of circulating nutrient fuels. Of the signals generated by mitochondria, ATP production is recognized as the principle nucleotide messenger for coupling mitochondrial metabolism with insulin secretion [1,2]. The postprandial increase in plasma glucose levels is matched by increased glucose uptake, glycolysis, and mitochondrial metabolism by the β-cell. Higher rates of glycolysis and mitochondrial metabolism lead to higher ATP levels triggering insulin release through a cascade of events mediated by the initial closure of the ATP-dependent potassium channels (KATP) and subsequent influx of Ca2+ [1-3]. In addition to ATP, rates of insulin secretion have been shown to be regulated by rates of mitochondria anaplerosis and mitochondrial-cytosolic substrate cycling [4-7]. This review will first discuss potential mechanisms regulating the activation of the mitochondria, followed by a discussion of the hypothesis that mitochondrial activation generates an integrated signal reflecting rates of oxidativephosphorylation, Kreb's cycle flux, anaplerosis, and substrate cycles that ultimately determines the rate of insulin exocytosis.
UNCOUPLING AS A REGULATOR OF MITOCHONDRIAL ACTIVATION: UCP2 ACTIVATION AND H+ DEPENDENT MITOCHONDRIAL CARRIERS
Increased rates of ATP synthesis is a direct consequence of the activation of mithochondrial metabolism. The efficiency of ATP synthesis by mitochondrial oxidativephosphorylation is determined in part by the proportion of the total proton flux channeled through ATP synthase from the mitochondrial intermembrane space back into the matrix. Any proton "leakage" across the inner mitochondria would therefore reduce the efficiency of ATP production. Although, the biological function and activation of UCP2 remains a matter of active debate, both in vivo and in vitro evidence support its functioning as a conduit for dissipating the mitochondrial protonmotive [8,9]. This has led to the hypothesis, supported by the work of Zhang et al. [8], that eliminating UCP2 would enhance both mitochondrial activation and fuel-stimulated insulin secretion. Alternatively, a teleological argument can be made that the functional consequences of UCP2 activity is to prevent the damaging effects of excessive reactive oxygen species (ROS) production. In support of this concept is the work of Krauss et al. [9] showing that while ROS as a class is not an activator of UCP2, superoxide is a potent activator. These results corroborate the recent report showing that the absence of UCP2 in several congenic strains of Ucp(-/-) mice have elevated markers of oxidative stress [10].
Our recent work exploring the mechanisms underlying impaired glucose-stimulated insulin secretion (GSIS) in patients with Maturity Onset of Diabetes of the Young-Type 3 (MODY-3) suggests that UCP2 activation may contribute to β-cell dysfunction [11]. Real-time 31P-NMR measured rates of ATP synthesis, showed that the mitochondria from β-cell lines expressing a dominant-negative mutation in the HNF-1α that is responsible for MODY-3 (DN-HNF-1α) were more uncoupled and less efficient for the synthesis of ATP under conditions of nutrient-stimulated insulin secretion [11]. Intriguingly, UCP2 mRNA expression was no different than the controls suggesting that enhanced activation of UCP2 is responsible for the change in uncoupling activity. Our observation is analogous to an earlier study examining the normalization of GSIS in phlorizin treated db/db mice, where islet function was enhanced, despite the persistence of high UCP2 expression in the islets [12]. While, they did not present data on mitochondrial function, the improved islet function may be explained in part by a reduction in the activation of UCP2.
Invoking UCP2 activation is only one of several possibilities though for diverting the protonmotive force away from ATP synthesis. The proton gradient is also used to couple the transport of metabolites and H+, leaving open the possibility that the apparent increase mitochondrial uncoupling is the result of increases in transport through SCL25 carriers that co-transport metabolites [13], or to increase flux through the H+/Ca2+ antiporter [14]. Further studies to identify the mechanism regulating mitochondrial uncoupling in β-cells, and its role in insulin secretion are needed to address these possibilities. Although, changes in the flux through the H+ dependent carriers may appear to be detrimental to mitochondrial function by reducing the efficiency of oxidative phosphorylation, they may in fact be necessary for other aspects of mitochondrial activation. Evidence to support this more holistic view of mitochondrial uncoupling is evident in the work of Wiederkehr et al. [15] on the role of mitochondrial alkalinization in mitochondrial activation.
MITOCHONDRIAL METABOLISM IS ACTIVATED BY MATRIX ALKALINIZATION
A quick review of the basic features of mitochondrial electrochemical potential will illustrate the notion that, like calcium, the alkalinization of mitochondrial matrix pH can be added as an activator of mitochondrial metabolism and hence fuel-stimulated insulin secretion [15]. Oxidation of pyruvate by the reactions of the Kreb's cycle is paired to the transfer of reducing equivalents to the electron transport chain localized within the inner mitochondrial membrane. A portion of the free energy lost during the transfer of electrons from each of the complexes of the electron transport chain is used to drive the ejection of protons from the matrix to the intermembrane space and sets up an electrical potential, Δψ, and a proton gradient, ΔpH, across the inner mitochondrial membrane. This electrochemical potential, ΔµH+, is used to drive the synthesis of ATP and the transport of various metabolites. Under normal physiological conditions, the Δψ is the major contributor to the overall magnitude of electrochemical potential contributing to ATP synthesis [16].
As mentioned above, the proton gradient, ΔpH, is utilized by several mitochondrial carrier proteins to transport metabolites across the mitochondria inner membrane [13,17], such that the proton gradient, per se, may contribute to the ability of the pancreatic β-cell to function as a nutrient sensor. Glucose stimulation increased the mitochondrial pH by ~0.4 in both primary β-cells and stable β-cell lines [15]. Consequently, ATP synthesis was dependent on the pH gradient with cellular ATP concentrations correlating with the kinetics of matrix alkalinization. The dependence of ATP synthesis upon matrix alkalinization to establish a pH gradient, together with the observation that alkalinization neither required nor was dependent upon Ca2+, suggest that fuel-stimulated mitochondrial alkalinization is another mechanism for activation of β-cell mitochondria. By regulating the transport of Ca2+ [14,18], and essential substrates such as phosphate, glutamate, and possibly pyruvate into the mitochondria [13,16], changes in pH gradient may regulate and sustain mitochondrial ATP synthesis and the production and transport of second messengers necessary for the amplification pathways of insulin secretion [1,4-7,16]. While the data clearly show a correlation between ATP synthesis and mitochondrial alkalinization, it is not yet clear which mitochondrial carriers, in addition to glutamate carrier 1, the phosphate carrier [13], and the recently identified Ca2+/H+ antiporter [14], may be affected as ΔpH increases, or how this impacts transport activity and the exchange of metabolites between the mitochondrial matrix and the cytosol.
ANAPLEROSIS AND MITOCHONDRIAL-CYTOSOLIC PYRUVATE CYCLING
The last several years have seen renewed interest in the role that substrate cycling of anaplerotic products may have for regulating insulin secretion. Unlike other metabolically-responsive cells, such as myocytes and hepatocytes, β-cells cannot accommodate the postpandrial increase in glucose uptake and glycolytic flux by storing the excess glucose as glycogen or eliminate it as lactate. Without an increase in energy demand that matches the increased glycolytic flux, feedback inhibition would limit the requisite metabolic responsiveness of the β-cell. Substrate cycling provides a mechanism to allow glycolytic and mitochondrial fluxes to increase in direct proportion to circulating concentrations of glucose and other fuel secretagogues.
Anaplerosis increases the concentrations of several Kreb's cycle intermediates generating a surplus of mitochondrial malate and citrate that can then be exported to the cytosol (Fig. 1). The concept that a β-cell pyruvate-malate cycle serves to couple metabolism with insulin secretion was advanced to provide a rationale for the relatively high activity of both pyruvate carboxylase and cytosolic malic enzyme in the β-cell [4-7,19,20]. Malate that is exported from the mitochondria to the cytosol is regenerated to pyruvate by cytosolic malic enzyme for cycling back to the mitochondria. As part of this pyruvate cycle, mitochondrial NADH reducing equivalents are transferred to cytosolic NADPH. Several lines of evidence support the notion of cytosolic NADPH assuming the role of a second messenger to couple increased mitochondrial activity with downstream cytosolic events leading to insulin secretion [21-23]. A more elaborate malate-pyruvate cycle occurs by way of citrate efflux via the citrate/isocitrate carrier. Together with cytosolic malic enzyme, ATP citrate lyase, and malate dehydrogenase, this pathway provide another means of cycling citrate to malate and back to pyruvate [24,25]. In addition to the fulfilling the task of generating the putative second messenger, cytosolic NADPH, Farfari et al. [5] suggested that this citrate-malate-pyruvate cycle serves to regenerate NAD+ and maintain glycolytic flux. Alternatively, cytosolic NADPH can be generated by a shunting of citrate to isocitrate and back to α-ketoglutarate.
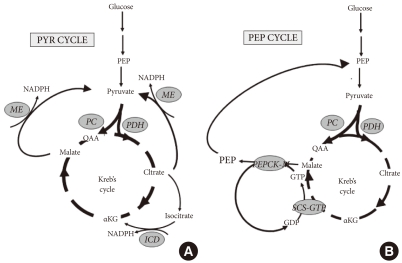
Pyruvate (PYR) and Phospho-enol-pyruvate (PEP) cycles are hypothesized to play a role in coupling anaplerosis through pyruvate carboxylase (PC), and by the rate of the Krebs cycle determined by pyruvate dehydrogenase (PDH). (A) Pyruvate cycling is dependent upon anaplerosis to carboxylate pyruvate to oxaloacetate (OAA) by PC, and thereby generate excess malate and citrate. Export of either transfers reducing equivalents from mitochondrial NADH to cytosolic NADPH by the action of malic enzyme (ME) [19]. Alternatively, citrate can be converted to isocitrate and shunted back toα ketoglutarate (αKG) by the activity of isocitrate dehydrogenase (ICD) and reduction of NADP+ to NADPH [24,25]. (B) PEP cycling is dependent upon the synthesis of GTP by the activity of the GTP isoform of succinyl-CoA-synthetase (SCS-GTP) [32]. The GTP synthesized by the activity of SCS-GTP is then used to drive the synthesis of PEP by the activity of the mitochondrial isoform of Phospho-enol-pyruvate carboxykinase (PEPCK-M) [33]. Mitochondrial PEP is exported to the cytosolic PEP pool to enhance glucose-stimulated insulin secretion by an undetermined mechanism.
The relevance of the pyruvate-malate cycle mediated by cytosolic malic enzyme has been challenged by recent results that confirmed a role for malic enzyme in INS-1 cells, but not rat islets [26]. However, the interpretation of the negative results when malic enzyme was silenced by adenovirus has to be accepted with reservation. The possibility of significant effects on GSIS from the control shRNA by itself could lead to misinterpretation of the observed changes in GSIS when using the shRNA to target malic enzyme. In addition, no supporting functional data was presented to support changes in enzyme activity or pyruvate cycling or redox state with the adenovirus-shRNA knock-down of malic enzyme. Thus, the role of malic enzyme and pyruvate cycling in metabolism-coupled insulin secretion remains an open question. Another possible, and likely explanation, is that there is a redundancy of these mitochondrial-cytosolic substrate cycle, and that malate-pyruvate cycle and the citrate-malate-pyrvate cycle work concurrently. The loss of any one cycle with low degree of metabolic control [27], as was done in the shRNA knock-down studies in islets [28], would lead to the conclusion that that particular cycle was unimportant for regulating glucose stimulated insulin secretion. A redundancy of cycles would share responsibility for the transfer of reducing equivalents to the cytosol, and could result in no single cycle having a high degree of metabolic control [27,28].
Regardless of the pathway, or pathways, responsible for providing cytosolic NADPH, compelling data supports its role as a mitochondrial second messenger. Two key steps in metabolism-coupled insulin secretion have been identified as sites where NADPH may exert regulatory control over rates of insulin release. The first site influences the initial events whereby NADPH levels can modulate intracellular concentrations of Ca2+. The second site of action is near the final events of secretion with NADPH playing a role in insulin granule docking and exocytosis. Accumulating evidence suggests that NADPH positively modulates Ca2+ influx through its binding to the regulatory subunit of voltage-dependent K+ (Kv) channels [29]. Kv channels act to repolarize the β-cell following the depolarization induced by closure of the KATP channels, and hence are negative regulators of insulin release. Additional levels of localized regulatory control over NADPH binding to Kv channels, and hence Ca2+ levels, are provided by the oxidoreductase activity of Kv subunits, and by other membrane associated oxidoreductases. Closer to the final events of insulin release, the reduction of glutathione by NADPH is hypothesized to be instrumental in potentiating insulin exocytosis [23]. Glutathione is thought to play a role in the post-translational modification of the t-SNARE proteins integral to insulin granule maturation and membrane docking. Hence, at least two mechanisms exist whereby NADPH, generated by pyruvate cycling, acts to regulate rates of insulin secretion in proportion to rates of mitochondrial metabolism.
OTHER NOVEL MITOCHONDRIAL SUBSTRATE CYCLES
In addition to mitochondrial-cytosolic malate cycling, two mammalian mitochondrial isoforms exist; malic enzyme 2 (ME2) with a preference for NAD+, and an NADP+-dependent malic enzyme 3 (ME3). In human and rat islets, and in INS-1 832/13 cells, ME2 is expressed at levels comparable to the expression of the cytosolic isoform, ME1. The enzymatic properties of ME2 support the hypothesis that ME2 can provide an alternative source of pyruvate from fumarate precursors such as glutamine when glycolytic flux is low [30]. Intriguingly, the work of Teller et al. [31] suggests that hetero-enzyme interactions of ME2 and the PDH complex serve to channel ME2-generated pyruvate to PDH in preference to the pool of free pyruvate. Using siRNA knock-down and isotope tracer technology, we found that reduction in ME2 blunted both glutamate flux into the Kreb's cycle and amino acid stimulated insulin secretion [19]. Thus, mitochondrial malic enzyme 2 mediates a mitochondrial-pyruvate cycle that utilizes the anaplerotic entry of glutamate to provide a source of pyruvate for mitochondrial activation.
Recently, a novel role for mitochondrial GTP production as a direct link to Kreb's cycle flux and glucose-stimulated insulin secretion has become recognized [32]. Mitochondrial GTP is an isolated pool and is generated in direct proportion to Kreb's cycle flux rate by the action of the GTP-dependent isoform of succinyl-CoA synthetase. Although isolated, the GTP that is generated supports the cycling of mitochondrial oxaloacetate to cytosolic phospho-enol-pyruvate (PEP) by the action of mitochondrial PEPCK (PEPCK-M) [33]. The existence of this cycle in β-cells (Fig. 1) had long been discounted, and the role of PEPCK-M in metabolism-coupled insulin secretion was unappreciated before these recent studies. In fact, mitochondrial PEPCK contributes a substantial fraction of cytosolic PEP (30% to 40%) in INS-1 cells and in rat islets, and the loss of this contribution almost completely blocks GSIS. Mitochondrial PEPCK, as well as GTP-SCS is expressed at substantial levels in human, mouse, and rat islets, supporting its importance across species, including humans. Preliminary data from our laboratory found that expression of SCS-GTP and is decreased in the DN-HNF-1α β-cell line, and warrant future studies on the possibility that dysregulation of the PEP cycling mediated by mitochondrial PEPCK may also contribute to impaired GSIS in MODY-3, and potentially type 2 diabetes metabolism.
RELEVANCE TO HUMAN ISLET BIOLOLGY
Mechanistic insights learned from our studies in β-cell lines, and mouse and rat islets are invaluable to better understanding the role of anaplerosis and substrate cycling in porcine and human islets. However, the multiplicity of isoforms involved in redundant and complementary substrate cycles, the variance in their degree of metabolic control, and the potential for species-dependency of expression and activity in islet β-cells make it critical to understand their functions in human islets. Using real-time PCR, we found that the human islets expressed all the critical pathways of anaplerosis, malate-pyruvate cycling and PEP cycling [34]. Pyruvate carboxylase, both the cytosolic and mitochondrial isoforms of malic enzyme, and both the ATP and GTP isoforms of succinyl-CoA synthetase, and mitochondrial PEPCK were expressed at similar levels. We also evaluated human islet β-cell function and found that with glucose-stimulation of insulin secretion, rates of ATP production, and the generation of the anaplerotic products, citrate and malate, were similar to those that we previously found in INS-1 832/13 cells and in rodent islets.
A recent report by MacDonald et al. [35] indicates a lesser role for pyruvate carboxylase and anaplerosis in human regulating stimulated insulin secretion. Multiple lines of evidence, including enzyme activities, mRNA expression, and substrate flux measurements, indicate that PC activity is reduced by as much as 80% to 90%. Despite the dramatic reduction in PC enzyme activity though, they found that the production of malate by human islets was only reduced by one-half in comparison to rat islets. Since malate production is dependent upon PC flux, the ample production of malate suggests that PC flux is still sufficient to support those cycles tied to insulin secretion. As is the case for rat islets, the enzyme distribution indicates a redundancy of pyruvate cycles in human islets. While, ATP Citrate Lyase, an enzyme integral to the citrate-malate-pyruvate cycle was found to be decreased, cytosolic malic enzyme activity and expression in human islets were similar to that in rat islets. In summary, the cumulative data suggests that the differences between rodent and human β-cells for the regulatory role of mitochondrial activation, anaplerosis, and substrate cycling may be one of degree, but not of function, for stimulated insulin secretion.
ARE OUR MECHANISTIC MODELS RELEVANT TO GSIS IN VIVO?
Finally, let us consider the possibility that our models are artifacts of our experimental conditions. The majority of our mechanistic models of metabolism-coupled insulin secretion are derived from results obtained under simplistic experimental conditions, and may not be representative of the complex in vivo conditions experienced by the islet β-cell within the pancreas. Consider the consensus theory of glucose-stimulated insulin secretion [1]. Central to this mechanism is that an increase in glucose metabolism and mitochondrial oxidative phosphorylation leads to an increase in cytosolic ATP concentrations to initiate closure of the KATP channel. This theory would predict that with the increase in oxidative phosphorylation, a proportional increase in oxygen consumption rates should occur. And in fact, this does hold when the glucose is increased from "basal" to stimulatory concentrations in minimal media. However, when the same studies are done in a more physiologically-complex media, the correlation between the change in oxygen consumption rate (OCR) and stimulated insulin secretion breaks down. In mouse βHC9 cells, Papas and Jarema [36] observed the change in the insulin secretion was proportional to the change in OCR, when the cells were stimulated with 15 mM glucose in the minimal media of phosphate buffered saline. However, despite a 2-fold increase in insulin secretion in response to 15 mM glucose, there was no observed change in OCR, and presumably oxidative phosphorylation, in more nutrient-complex Dubelco's Minimal Eagle Media (DMEM) buffer. We observed similar results for the rat insulinoma INS-1 832/13 cells, and for isolated rat islets.
Studies to identify which of the several possible scenarios could be responsible for this anomaly are under active investigation [37]. Using 31P-NMR to measure ATP synthesis in real-time, we were able to show that increasing glucose from 2.5 mM to 15 mM led to identical increases in ATP synthesis rates in both minimal and complex media, with the correlation of insulin secretion rates vs. ATP synthesis rates falling on two distinct and approximately parallel lines for KRB and DMEM. Another possibility is that the complexity of the media perturbs the role of anaplerosis and the export of putative mitochondria second messenger. Using [U-13C]glucose as a tracer to calculate the relative pathways and substrates into the Kreb's cycle. In both DMEM and KRB, the transition from basal concentrations to stimulatory concentrations led to similar increases in the entry of glucose into the Kreb's cycle following its conversion to pyruvate and acetyl-CoA for citrate synthesis. Increasing glucose led to similar changes in anaplerotic flux, such that insulin secretion rates strongly correlated with anaplerosis via pyruvate carboxylase flux. In fact, the correlation in KRB was congruent that the correlation in DMEM. The strong correlation between anaplerosis and rates of insulin secretion in DMEM and KRB, and especially their congruency, lends further support to the hypothesis that the export of mitochondrial second messengers couples mitochondrial metabolism to insulin secretion. As discussed above, NADPH is a leading candidate for such a role; however the studies of Kibbey et al. and Stark et al. [32,33] hint at the possibility that other unidentified cytosolic signaling pathways coupled to metabolism augment insulin secretion in vivo.
In conclusion, mitochondria of pancreatic islet β-cells regulate insulin secretion by means of an integrated signal provided by ATP production, anaplerosis, mitochondrial-cytosolic and mitochondrial-mitochondrial substrate cycling, the regulation of cellular redox state, and the production of other secondary signaling intermediates. Those signaling mechanisms that are linked to the rates of anaplerosis, the Kreb's cycle, and substrate cycling enhance regulated rates of insulin secretion above that provided by ATP production alone.
ACKNOWLEDGMENTS
The author thanks Rebecca L. Pongratz for the excellent graphics work.
Notes
No potential conflict of interest relevant to this article was reported.