Roles of Reactive Oxygen Species on Insulin Resistance in Adipose Tissue
Article information

Abstract
Obesity resulting from the delivery of an excess amount of energy to adipose tissue from glucose or free fatty acids is associated with insulin resistance and adipose tissue inflammation. Reactive oxygen species (ROS) have been implicated as contributors to both the onset and the progression of insulin resistance. ROS can be generated by overloading the mitochondrial oxidative phosphorylation system, and also by nicotinamide adenine dinucleotide phosphate oxidases (NOX) produced by either adipocytes, which only produce NOX4, or by macrophages, which produce mainly NOX2. The source of the ROS might differ in the early, intermediate and late stages of obesity, switching from NOX4-dependence in the early phases to NOX2-dependence, in the intermediate phase, and transiting to mitochondria-dependence later in the time course of obesity. Thus, depending on the stage of obesity, ROS can be generated by three distinct mechanisms: i.e., NOX4, NOX2, and mitochondria. In this review, we will discuss whether NOX4-, NOX2-, and/or mitochondria-derived ROS is/are causal in the onset of adipocyte insulin resistance as obesity progresses. Moreover, we will review the pathophysiological roles of NOX4, NOX2, and mitochondria-derived ROS on adipose tissue inflammation.
INTRODUCTION
Many lines of evidence support the proposal that visceral obesity is strongly associated with features of the metabolic/insulin resistance syndrome, and that obesity predicts the development of both type 2 diabetes and cardiovascular disease (CVD) [123]. Obesity is associated with a chronic low-grade inflammation [456], as evidenced by an increase in circulating inflammatory markers, such as C-reactive protein, serum amyloid A (SAA), and interleukin 6 [789]. The presence of systemic inflammation in visceral obesity has been linked to an increased risk of developing CVD and type 2 diabetes [101112]. Obesity results when there is an imbalance between energy ingested and energy expended [13]. A relative excess of energy (either genetic or diet-induced) results in two major cellular features; adipocyte expansion and infiltration of inflammatory cells into adipose tissue in both mice and humans [141516]. Adipocytes and macrophages both generate inflammatory molecules, which lead to insulin resistance and systemic inflammation. Certain saturated free fatty acids (SFAs; laurate, myristate, and palmitate) increase inflammatory genes in adipocytes [17]. These events are associated with the generation of reactive oxygen species (ROS) and nuclear factor κB (NF-κB) transactivation. Excess glucose and/or certain SFAs increased ROS generation and NF-κB translocation [17]. Treatment with the antioxidants, N-acetyl cysteine, catalase and superoxide dismutase (SOD) repressed ROS generation and NF-κB translocation stimulated by excess glucose 4tate, and decreased inflammatory gene expression [17]. Thus, glucose- and palmitate-stimulated ROS generation appears to play an important role in adipocyte inflammation. Although several mechanisms such as endoplasmic reticulum (ER) stress, hypoxia, and adipocyte death have been reported to be related to adipocyte inflammation during obesity [1819], ROS generation is upstream of ER stress and apoptosis of adipocyte occurs at later stages of obesity [1820]. ROS is a key-modulator that activates the initial the sequence of events that leads to adipose tissue inflammation. The relative contribution of ROS derived from different sources in adipocytes during the progression of obesity remains unknown. Therefore, as a first step, our review has been chosen to focus on the role of various sources of ROS generated in adipose tissue on inflammation and insulin resistance.
ROS GENERATION AND OBESITY
Chronic ROS production has recently been suggested to be an important contributor to the pathogenesis of obesity-associated insulin resistance [2122]. Physiologically, ROS is required for differentiation of adipocytes [2324]. These physiological ROS exist for short time and are well regulated by insulin signaling. Our review focuses on the pathophysiological roles of ROS in adipocytes inflammation; i.e., the effects of prolonged ROS generation by excess nutrients and the effect of ROS unregulated by insulin signaling. ROS in visceral adipose tissue are significantly increased in genetically obese mice and mice made obese by consumption of a high-fat diet [25]. The direct relationship between ROS generation and obesity is still not entirely clear. ROS can be generated by adipocytes during metabolism of excess nutrients. ROS also can be generated by macrophages, which accumulate in adipose tissue in obesity. Therefore, it is important to understand the source of ROS during the progression of obesity. In the early insulin sensitive state, energy flux from nutrient excess flows into lipogenesis, in which excess glucose and free fatty acid (FFA) are used for triglyceride synthesis by adipocytes. In the insulin sensitive state, insulin activates the insulin receptor tyrosine kinase which leads to stimulation of insulin receptor substrate proteins and phosphatidylinositol 3-kinase (PI3-kinase) [26]. Next, PI3-kinase activates downstream protein kinases, including Akt/protein kinase B [2627]. Consequently, these events increase intracellular glucose uptake into adipocytes via insulin-stimulated translocation of glucose transporter 4 molecules to the cell membrane [27]. In this state, FFA are stored as triglyceride by activating the production of proteins involved in lipid metabolism/uptake, including lipoprotein lipase, fatty acid transport protein, and acetyl CoA-synthase [28]. However, it is totally unknown whether these ROS casually trigger the insulin resistance, or are a casual bystander of insulin resistance in adipocytes. Thus, although much is known about energy flux in adipocytes during the progression of obesity, little is known as to whether this energy flux leads to differences in the generation of ROS in the early and late stages of obesity, nor is there a good understanding of the potential pathophysiological roles of ROS derived in these stages.
ROS AND NOX4 IN THE EARLY STAGES OF OBESITY
We previously have shown that excess glucose and palmitate are not metabolized to a major extent via mitochondrial oxidation. Instead excess nutrients activate nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOX) [29]. Members of the NOX family are membrane-bound enzyme complexes that transfer electrons from NADPH to oxygen, generating superoxide. This short lived, non-membrane permeable ROS is converted, to a longer lived membrane permeable ROS, hydrogen peroxide by SOD [30] or spontaneously. The NOX family has seven isoforms, NOX1, NOX2, NOX3, NOX4, NOX5, dual oxidase 1 (DUOX1), and DUOX2, which are strongly conserved in mammals and are widely expressed in various tissues [31]. NOX1 to 3 require additional cytosolic activators (p47phox, p67phox, NADPH oxidase organizer 1) whereas NOX4 is constitutively active and independent of an activator protein (AP) [313233]. NOX5 and DUOX1/2 need intracellular calcium to activate ROS generation [3435]. It is therefore assumed that NOX1 to 3 mediates short-term effects, while NOX4 is responsible for long term effects. We have found that NOX4 is the major NOX isoform in cultured murine and human adipocytes [29]. Moreover, silencing NOX4 decreased ROS generation stimulated by excess glucose as well as palmitate, leading to inhibition of monocyte chemoattractant protein-1 and SAA3 expression in vitro [29]. Thus, NOX4-derived ROS may be a common mediator induced by both excess glucose and palmitate in adipocytes (Fig. 1). Other studies showed that NOX4 activity increases in the adipose tissue with diet-induced obesity (DIO), and NOX inhibitor, apocynin treatment reduces ROS generation [25]. However, whether NOX4-derived ROS itself can promote the onset of insulin resistance in adipocytes during the progression of obesity is unknown and needs to be investigated.
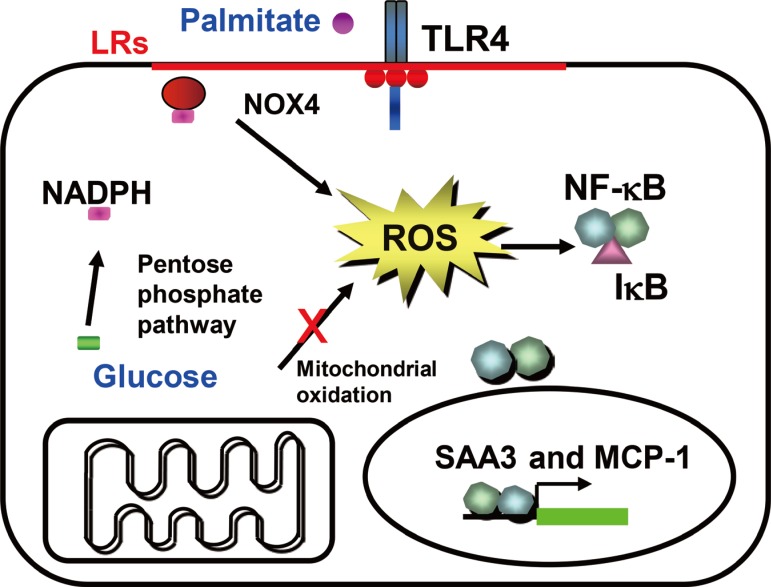
Hypothesis for a mechanism by which excess glucose and saturated free fatty acids (SFAs) affect reactive oxygen species (ROS) generation in adipocytes in early stages of obesity. Excess glucose generates ROS via the pentose phosphate pathway, rather than by overloading mitochondrial oxidative phosphorylation, while SFA generate ROS following activating, Toll-like receptor 4 (TLR4) or lipid rafts. Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 4 (NOX4) might be the common mediator of ROS generation by both excess glucose and SFAs. ROS generated by both glucose and SFA activate nuclear factor κB (NF-κB). LR, lipid raft; SAA3, serum amyloid A3; MCP-1, monocyte chemoattractant protein-1.
The pentose phosphate pathway (PPP) generates NADPH and pentose from the 6 carbon glucose and is a major source of cellular NADPH. Glucose-6 phosphate dehydrogenase (G6PD) is the rate-limiting enzyme in the PPP. Recent studies indicate that G6PD expression is upregulated in adipose tissue in genetic and DIO, and that its overexpression is associated with increased adipocyte inflammation and ROS generation [3637]. Other studies have shown that treatment with dehydroepiandrosterone (DHEA), a G6PD inhibitor, reduces obesity in Zucker diabetic fatty rats [38]. We found that NADPH content and PPP activity were increased by excess glucose, but not by palmitate in 3T3-L1 adipocytes [29]. Moreover, G6PD inhibitors, DHEA and 6-aminonicotinamide, or silencing G6PD all inhibited ROS generation and monocyte chemotactic factor gene expression by both high glucose and palmitate in 3T3-L1 adipocytes [29]. These studies support the concept that PPP and G6PD could be modulators or mediators of adipose tissue inflammation (Fig. 1). During the initial stage of energy access, we hypothesize that adipocytes will continue to actively store triglycerides derived from excess nutrients, and will demonstrate increased PPP activity and NADPH content, which lead to NOX4-derived ROS generation (Fig. 1).
However, whole body NOX4 deficiency has been reported to worsen adipose tissue inflammation in a model of DIO in mice [39]. Since NOX4 activity is essential for pre-adipocyte differentiation to adipocytes [23], blunted adipogenesis in the absence of NOX4 would reduce the number of adipocytes, allowing the remaining adipocytes to become more hypertrophic, thereby leading to adipose tissue inflammation. Indeed, expression of adipogenesis genes (peroxisome proliferator-activated receptor γ [PPARγ] and CCAAT-enhancer-binding protein α [C/EBPα]) was inhibited in whole body NOX4 knockout mice [39]. Therefore, it is imperative to investigate the alteration of NOX4 activity in mice during the pathophysiological progression of obesity where adipogenesis is intact, and to study the effect of adipocyte-specific deficiency of NOX4.
ROS AND NOX2 IN THE INTERMEDIATE STAGES OF OBESITY
Another potentially important source of ROS in obesity is from macrophages that are recruited and accumulate in obese adipose tissue. Obesity provokes changes in T-cell subsets and increases the infiltration and accumulation of activated macrophages in adipose tissue [404142]. These recruited and activated immune cells can promote the generation of ROS by NOX2, which is predominately expressed activated in T-cells and macrophages [43]. Immune cells in the inflammatory environment created by obesity could generate NOX2-derived ROS. Whole body deficiency of NOX2 shows attenuation of adipose tissue inflammation and insulin resistance in mice fed a high fat diet [44]. This implies that NOX2 from immune cells may play a role in adipose tissue inflammation in the intermediate stages of obesity. However, whether these NOX2-dervied ROS itself has effects in adipose tissue inflammation and can promote the onset of insulin resistance in adipocytes during the progression of obesity is unknown and needs to be investigated.
ROS AND MITOCHONDRIA IN THE LATE STAGES OF OBESITY
The mitochondrial electron transport chain is a primary site for ROS production. ROS generation is accelerated when the flow of electrons through the electron transport chain is overload, resulting in leakage of electrons. Overloaded mitochondria mainly produce superoxide, which can signal intracellularly, leading to a pro-inflammatory signaling cascade that includes NF-κB and AP-1 activation [4546]. Although glucose excess presumably drives the overflow of electrons in mitochondria, other energy sources in adipocytes are preferred in the late stages of obesity since uptake of glucose is limited by the insulin resistance of adipocytes. When glucose consumption is reduced by insulin resistance, adipocytes start to use FFA from triglyceride stores for energy (Fig. 2). This alteration of energy flux into β-oxidation could overwhelm the capacity of mitochondria, leading to leakage of electrons (Fig. 2).

Hypothesis for a mechanism by which mitochondrial β-oxidation of free fatty acid (FFA) from triglyceride stores affects reactive oxygen species (ROS) generation in adipocytes in the late stage of obesity. Uptake of excess glucose and FAA are inhibited, resulting in a decrease of pentose phosphate pathway and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 4 (NOX4) activity. FFA from fat stores overload mitochondrial oxidative phosphorylation and generate ROS. Mitochondria-derived ROS activate nuclear factor κB. IR, insulin receptor; IRS, insulin receptor substrate; GLUT4, glucose transporter 4; LPL, lipoprotein lipase; FATP, fatty acid transport protein; HSL, hormone-sensitive lipase.
When pro-inflammatory macrophages accumulate in adipose tissue as a result of the NOX4-mediated generation of macrophage chemoattractants by adipocytes, the inflammatory environment induced by these macrophages will be expected to result in adipocyte insulin resistance [47]. With the advent of sufficient insulin resistance, adipocytes will stop storing additional triglycerides, will utilize stored fatty acids from triglycerides, and will show evidence of decreased PPP activity and NADPH content. At such times, adipocyte-derived NOX4 might have a limited role in ROS generation and adipose tissue inflammation. Instead, mitochondria will now take the place of NOX4 and play a pivotal role in ROS generation and adipose tissue inflammation (Fig. 2). In support of this hypothesis are studies that show the production of pro-inflammatory cytokines and macrophage chemoattractants by adipocytes at an early time point (1 week), while adipose tissue insulin resistance occurs much later (after 14 weeks) during the development of DIO in mice [4849]. These studies also show that even though some infiltration of macrophages occur at early time points, massive infiltration of macrophage happens much later (after 12 weeks) [49]. Thus, these studies strongly suggest that adipocytes become insulin resistant during the later stages of obesity, while adipocytes are insulin sensitive during the early stages of obesity. Recent studies also have reported that fasting and caloric restriction in mice in which insulin signaling is disturbed and β-oxidation of FFA is increased in adipocytes, results in the generation of chemoattractants and an increase of macrophage accumulation in adipose tissue [5051]. These studies support that mitochondria and β-oxidation of fatty acids might lead to ROS generation. Therefore, we hypothesize that mitochondria-derived ROS accounts for massive macrophage accumulation, worsening insulin resistance and systemic inflammation during the late stages of obesity (Fig. 2). Also, we hypothesize that a temporal transition of the source of ROS between NOX4 and mitochondria as obesity progresses is responsible for conversion to a more insulin resistant phase of obesity.
CONCLUSIONS
An overall role of distinct sources of ROS for adipose tissue inflammation is that (1) in the early stages of obesity, NOX4-derived ROS from adipocytes provoke the onset of insulin resistance and initiates the recruitment of immune cells in adipose tissue, (2) in the intermediate stages of obesity, NOX2-derived ROS from infiltrated immune cells worsens adipocyte insulin resistance and adipose tissue inflammation, and (3) in the late stages of obesity, mitochondria-derived ROS from adipocytes maintain the adipose tissue inflammation and insulin resistance. We summarize how these three distinct sources of ROS might affect adipocyte insulin resistance and adipose tissue inflammation in the early, intermediate, and late stages of obesity (Fig. 3).

Scheme of all mechanisms by which these three distinct sources of reactive oxygen species (ROS) might affect adipocyte insulin resistance and adipose tissue inflammation. NOX, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase.
Several large clinical trials have failed to show any beneficial effects of consumption of antioxidant supplements in the prevention of insulin resistance [52535455]. Nevertheless, from studies in obese mice there is increasing evidence that antioxidant might be beneficial in attenuating insulin resistance and restoring insulin signaling. For example, the antioxidant manganese tetrakis porphyrin and the cell permeable small-peptide antioxidant SS31 (D-Arg-2',6'-dimethyltyrosine-Lys-Phe-NH2) improve insulin sensitivity without altering body weight in a genetic and DIO mice [4356]. Moreover, transgenic mice overexpressing SOD2 exhibit improvements in glucose tolerance and insulin sensitivity resulting from consumption of a high fat diet [57]. A discrepancy between basic research and clinical studies may be due to the fact that we have yet to address the importance of the timing of ROS generation, the source of ROS, and tissue specific effects of ROS. Findings out these discrepancies would be likely to have important translational implications related to the development of antioxidants targeting NOX4- or mitochondria-derived ROS in different stages of obesity.
ACKNOWLEDGMENTS
This work was supported in part by National Institutes of Health grants HL092969, 094352, DK-035816, and a Beginning Grant-in-Aid from the American Heart Association.
Notes
CONFLICTS OF INTEREST: No potential conflict of interest relevant to this article was reported.