- Current
- Browse
- Collections
-
For contributors
- For Authors
- Instructions to authors
- Article processing charge
- e-submission
- For Reviewers
- Instructions for reviewers
- How to become a reviewer
- Best reviewers
- For Readers
- Readership
- Subscription
- Permission guidelines
- About
- Editorial policy
Articles
- Page Path
- HOME > Diabetes Metab J > Volume 35(4); 2011 > Article
-
ReviewPolymeric Gene Delivery for Diabetic Treatment
- Sung Wan Kim1,2
-
Diabetes & Metabolism Journal 2011;35(4):317-326.
DOI: https://doi.org/10.4093/dmj.2011.35.4.317
Published online: August 31, 2011
1Department of Pharmaceutics and Pharmaceutical Chemistry and Department of Bioengineering, University of Utah, Salt Lake City, UT, USA.
2Department of Bioengineering, Hanyang University, Seoul, Korea.
- Corresponding author: Sung Wan Kim. Department of Pharmaceutics and Pharmaceutical Chemistry and Department of Bioengineering, University of Utah, Salt Lake City, UT 84112, USA. SW.Kim@pharm.utah.edu
Copyright © 2011 Korean Diabetes Association
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- ABSTRACT
- INTRODUCTION
- DELIVERY OF IL-10 PLASMID DNA TO PREVENT AUTOIMMUNE INSULITIS OF NOD MOUSE
- REPRESSION OF GAD AUTOCENTIGEN EXPRESSION IN PANCREATIC β-CELL BY DELIVERY OF ANTISENSE PLASMID
- GLP-1 GENE DELIVERY
- PREVENTION OF TYPE-1 DIABETES USING RAE-1 PLASMID AND EPHRIN CONJUGATED POLYETHYLENE GLYCOL TRIETHYLENE TETRA AMINE/CYSTAMINE BISABRYLAMIDE (EPH-PEG-TETA/CBA) COPOLYMER
- Fas siRNA DELIVERY IN CYCLOPHOSPHAMIDE INDUCED WITH DIABETES IN NOD MICE
- ACKNOWLEDGMENTS
- NOTES
- REFERENCES
ABSTRACT
- Several polymers were used to delivery genes to diabetic animals. Polyaminobutyl glycolic acid was utilized to deliver IL-10 plasmid DNA to prevent autoimmune insulitis of non-obese diabetic (NOD) mouse. Polyethylene glycol grafted polylysine was combined with antisense glutamic acid decarboxylase (GAD) MRNA to represent GAD autoantigene expression. GLP1 and TSTA (SP-EX4) were delivered by bioreducible polymer to stop diabetic progression. Fas siRNA delivery was carried out to treat diabetic NOD mice animal.
- Somatic gene therapy is defined as the introduction of genetic information into cells resulting in the production of proteins which correct or modulate a disease. Gene therapy is advantageous over conventional protein delivery because it is designed to overcome the limitations due to protein delivery such as low bioavailability, poor pharmacokinetics, and complex purification processes that result in high manufacturing costs, unstable biological activity, antibody formation, and toxicity. In addition, gene therapy can be used to restrict the expression of a therapeutic protein to specific cells within the body, to control expression levels, and achieve sustained expression over time as opposed to the "burst effect" which is the typical pharmacokinetic profile of protein delivery. There are 2 main types of gene therapy, viral and non-viral gene delivery. Viral-mediated gene therapy is based on the use of attenuated or defective viruses produced by genetic engineering and includes the use of retroviruses, adenoviruses, adeno-associated viruses, and herpes viruses. Viral vectors have much greater transfection efficiencies in comparison to non-viral vectors, however, there are many drawbacks including the induction of immunological responses, random insertion of viral sequences into the host chromosomes resulting in the activation of oncogenes or the inactivation of tumor suppressor genes, recombination events leading to virulent viral particles, limitations of the DNA size for encapsulation as well as large scale production of viral titers [1-5].
- Plasmid DNA, which encodes information for the expression of 1 or more therapeutic proteins, is highly negatively charged and has relatively high molecular weight. Hence, the intracellular transport of plasmid DNA is limited due to the charge repulsion between the plasmid DNA and negatively charged plasma membrane. In addition, plasmid DNA is rapidly cleared from the bloodstream due to uptake by the liver and degradation due to endonucleases [6]. These limitations can be overcome via the use of cationic polymer-based carriers that can be utilized to condense plasmid DNA molecules into particles protecting the plasmid DNA from the degradation by nucleases, resulting in delayed clearance time from the bloodstream, and promote internalization of complexes on cellular surfaces due to electrostatic interactions. The positive charges on the complexes enable stabilization in water or low concentration buffers. Systemic gene delivery requires the rational design of a polymeric gene vehicle with high solubility, low cytotoxicity, serum stability, efficient endosomal release, adequate protection against endonucleases, and most importantly, a high transfection efficiency and gene expression.
- Type 1 diabetes was formerly called insulin-dependent or juvenile onset diabetes. Type 1 diabetes accounts for 5% to 10% of all diabetes cases (17 million people) in the United States [7]. In a person with type 1 diabetes, autoimmune processes destroy the pancreatic β-cells, thus, no insulin is produced in the pancreas. Type 2 diabetes was formerly called non-insulin-dependent or adult-onset diabetes and arises due to insulin resistance in which the body cannot use insulin properly, combined with relative insulin deficiency. Type 2 diabetes typically presents in middle and late life, although an increasing number of children and teenagers around the world are developing type 2 diabetes due to an increase in obesity. Type 2 diabetes does not exhibit a clear set of symptoms. When fully developed, abnormalities in both insulin secretion and insulin action are present. Type 2 diabetes is characterized by an increase in basal glucose concentrations, upon which are superimposed exaggerated postprandial glucose excursions induced by a combination of β-cell dysfunction and impaired insulin sensitivity. Type 2 diabetes mellitus occurs when the pancreatic β-cells fail to generate an adequate insulin level to maintain euglycemia in the setting of insulin resistance. The molecular basis of insulin resistance and β-cell failure in type 2 diabetes remains unclear. Type 2 diabetes constitutes 90% to 95% of all diabetes cases, and the incidence of type 2 diabetes is rapidly increasing worldwide [8].
- Oral agents necessary to attain the goal of near-normal glycemia are needed. Treatment for type 2 diabetes relies on several approaches mainly to reduce the hyperglycemia [9]. The oldest class of oral antihyperglycemic agents is the sulfonylurea and remains the most commonly used initial pharmacologic treatment for the disease due to potency, effectiveness, and low cost. The sulfonylureas work by binding to receptors on the surface of pancreatic islet β-cells and stimulating the release of endogenous insulin [10]. However, sulfonylurea stimulation of insulin secretion is not glucose-dependent, and hence hypoglycemia is an adverse effect of the treatment. Another problem with sulfonylurea is that after an extensive treatment, many patients have shown secondary failure and no longer respond to sulfonylurea treatment [11]. There are several other therapies currently used for type 2 diabetes: metformin, which reduces hepatic glucose production, thiazolidinediones, which enhance insulin action and α-glucosidase inhibitors which interfere with gut glucose adsorption. These therapies also have significant side effects and have limited efficacy [12].
- Therefore, newer physiological approaches are desperately needed for treatment of type 2 diabetes. The use of hormones, other than insulin, is a relatively new area of clinical investigation and therapy for the treatment of diabetes. Preventing the risk of hypoglycemia by potentiating insulin secretion in a glucose-dependent manner is possible.
- In this manuscript, several works in the area of gene delivery, which have been performed at the University of Utah, are described.
INTRODUCTION
- Early in 2000, the author of the present study previously reported biodegradable poly [α-(4-aminobutyl)-L-glycolic acid] (PAGA) can protect and condense plasmid DNA from DNase I. Whether the systemic administration of pCAGGS mouse IL-10 (mIL-10) expression plasmid complexed with PAGA can reduce the development of insulitis in non-obese diabetic (NOD) mice was investigated. PAGA/mIL-10 plasmid complexes were stable for more than 60 minutes, although DNase I destroyed the naked DNA within 10 minutes. The PAGA/DNA complexes were injected into the tail vein of 3-week-old NOD mice. Serum mIL-10 level peaked at 5 days after injection, and could be detected for more than 9 weeks. The prevalence of severe insulitis on 12-week-old NOD mice was markedly reduced by the intravenous injection of PAGA/DNA complex (15.7%) compared with naked DNA injection (34.5%) and non-treated controls (90.9%). In conclusion, systemic administration of pCAGGS mIL-10 plasmid/PAGA complexes can reduce the severity of insulitis in NOD mice. The study shows the PAGA/DNA complex has the potential for the prevention of autoimmune diabetes mellitus [13].
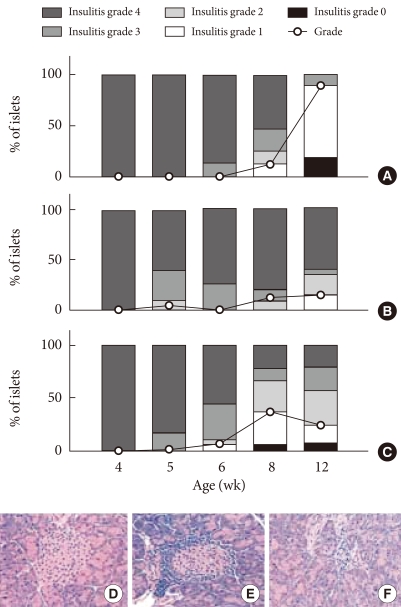
- Two hundred microliters of PAGA/plasmid complexes (2/1, +/-) were injected into the tail vein of NOD mice at a dose of 100 µg DNA per mouse. At designated times, the animals were killed by cervical dislocation and the blood was collected from the inferior vena cava, centrifuged at 3,500 rpm for 15 minutes and stored at -70℃ (n=2 in each group, total n=30). The pancreas and liver were also harvested. The amount of serum mIL-10 was determined by using an enzyme-linked immunosorbent assay (ELISA). For hematoxylin and eosin staining, the pancreas was fixed in 20% formalin containing 75 mm phosphate buffer at pH 7.0, and embedded in paraffin. Five-micron thick sections were cut and stained with hematoxylineosin. More than 20 islets from each pancreas were examined using double blind methods from 3 animal groups. To characterize the progression of insulitis, each islet was assigned by using the following insulitis grading system: 28 Grade 3 and 4 were considered as severe insulitis (grade 0, normal islets; 1, mononuclear cell infiltration largely in the periphery in less than 25% of the islet; 2, 25% to 50% of islet showing mononuclear infiltration; 3, over 50% of islet showing mononuclear infiltration; and 4, small retracted islet with few mononuclear cells); the results are shown in Fig. 1.
- In addition, a plasmid in which the expression of interleukin-4 (IL-4) was driven by the rat insulin promoter was constructed (pRIP-IL4). Water soluble lipopolymer (WSLP)-pRIP-IL4 complex was characterized by pancreas β-cell specific and glucose responsive expression of IL-4. pRIP-IL4 was completely retarded at a 6:1 or higher N/P (nitrogen atom of WSLP/phosphate of plasmid) ratio in 1% agarose gel. In addition, WSLP protected plasmid DNA from DNase I for more than 1 hour. In the cytotoxicity assay, WSLP showed less cytotoxicity than polyethylenimine (PEI) (25,000 Da) to mouse insulinoma (MIN6) cells. ELISA showed pRIP-IL4 expressed much higher levels of IL-4 in MIN6 cells than in NIH3T3 cells. The expression level of IL-4 by pRIP-IL4 increased with increasing concentration of glucose. Additionally, IL-4 was expressed in a dose-dependent manner. The WSLP-pRIP-IL4 system could be useful in the development of a pancreas-specific expression system for the prevention of diabetes without systemic side effects [14].
- Studies of animals with spontaneous autoimmune diabetes have revealed autoreactive T-cells that mediate islet β-cell destruction belong to the Th1 subset (producing IL-2 and IFN-γ), whereas regulatory T-cells are Th2 type (producing IL-4 and IL-10). The effect of combined delivery of plasmid DNA encoding IL-4 and IL-10 was evaluated using the degradable, cationic polymeric carrier, PAGA, in NOD mice. In the liver of NOD mice, mouse Il4 and Il10 mRNA was detected 5 days after intravenous injection of both PAGA-Il4 and PAGA-Il10 plasmid complexes. Six weeks after injection, 75% of observed islets were intact compared with less than 3% in the control group. Furthermore, in the treatment group, only 5% of the islets were severely infiltrated by the lymphocytes compared with over 30% in the control group. Glucose levels were measured weekly up to 32 weeks of age, revealing co-injection of PAGA-Il4 and PAGA-Il10 plasmids prevented the development of diabetes in 75% of the treated animals. Thus, combined administration of mouse IL4 and IL10 plasmids prevented the development of autoimmune diabetes in NOD mice [15].
DELIVERY OF IL-10 PLASMID DNA TO PREVENT AUTOIMMUNE INSULITIS OF NOD MOUSE
- Silencing the expression of glutamic acid decarboxylase (GAD) in transgenic NOD mice was reported to completely protect islet β-cells against the development of diabetes, suggesting the repression of GAD autoantigen by somatic gene delivery can prevent autoimmune destruction of pancreatic β-cells. To repress GAD expression in islet β-cells, an antisense GAD mRNA expression plasmid (pRIP-AS-GAD) was delivered using poly (ethylene glycol)-grafted poly-L-lysine (PEG-g-PLL) as a gene carrier. In a gel retardation assay, the pRIP-AS-GAD/PEG-g-PLL complex was completely retarded above a weight ratio of 1:1.5 (plasmid:PEG-g-PLL). PEG-g-PLL protected the plasmid DNA from DNase I for more than 60 minutes. In a reporter gene transfection assay, PEG-g-PLL showed the highest transfection efficiency at a weight ratio of 1:3. The pRIP-AS-GAD/PEG-g-PLL complex was also transfected into a GAD-producing MIN6 cell line. The antisense mRNA was expressed specifically in β-cells and expression was dependent on the glucose level. The repression of GAD after transfection of pRIP-AS-GAD was confirmed by immunoblot assay. In addition, in vivo expression of antisense RNA in the pancreas was confirmed by RT-PCR after intravenous injection of the complex into mice. Therefore, the study revealed the pRIP-AS-GAD/PEG-g-PLL system is applicable for the repression of GAD autoantigen expression [16].
- An islet cell targeting polymeric gene carrier was synthesized by conjugating an anti-GAD Fab' fragment to PEI via a PEG linker (PEI-PEG-Fab'). The Fab' fragment was prepared from a murine monoclonal antibody against GAD, which has been identified as a major auto-antigen expressed in islet cells and used as a targeting moiety for islet cell targeting. The elec trophoretic migration of plasmid DNA (pCMVLuc)/PEI-PEG-Fab' complexes in agarose gel was completely retarded above the N/P ratio of 2. The complexes demonstrated a size of 100 to 275 nm with an almost neutral surface charge. Confocal microscopy revealed the PEI-PEG-Fab' complexes showed much higher cellular binding and uptake efficiency compared to PEI-PEG complexes. The PEI-PEG-Fab' showed approximately 10-fold higher transfection efficiency (relative luciferase activity) than PEI-PEG in GAD-expressing MIN6. However, the transfection efficiency of PEI-PEG-Fab' reduced the PEI-PEG in GAD-negative cells (293) and in the presence of competitive free Fab's. Considering the neutral surface charge of the PEI-PEG-Fab' conjugate complexes with DNA and selectivity toward the islet cells expressing a specific antigen, PEI-PEG-Fab' conjugate could be considered as a potential candidate of the systemic gene therapy for the treatment of type I diabetes [17].
REPRESSION OF GAD AUTOCENTIGEN EXPRESSION IN PANCREATIC β-CELL BY DELIVERY OF ANTISENSE PLASMID
- In 1930, La Barre introduced the term 'incretin' to describe the activity of the gut that could enhance the endocrine secretion of pancreas [18]. In response to the ingestion of food, both insulin and insulin counter-regulatory hormones are released into the blood stream. Insulin secretion is related to the serum concentration of absorbed nutrients, such as glucose, amino acids, and fatty acids. Insulin secretion is also influenced by incretins. The incretins are intestinal hormones that augment insulin secretion in the presence of elevated glucose levels [19]. In humans, 2 gastrointestinal peptide hormones are thought to be responsible for the incretin effect [20]. One is glucose-dependent insulinotropic polypeptide (GIP), which was originally named 'gastric inhibitory polypeptide' [21]. As the cloning of cDNAs encoding the preproglucagons of the anglerfish was accomplished in the early 1980s, the second incretin hormone was discovered [22]. Shortly after cloning the proglucagon cDNAs of anglerfish, the proglucagon cDNAs of human and other mammals were cloned [23]. Glucagon-like peptide-1 (GLP-1) is synthesized in the intestinal L-cells by post-translational processing of the glucagon precursor, preproglucagon, and is released into the circulation in response to a meal. The GLP-1 isoforms GLP-1 (7-37) and GLP-1 (7-36) amide are well known as bioactive insulinotropic peptides derived from preproglucagon. Studies of the secretion of GLP-1 and GIP in type 2 diabetic patients have shown that meal-induced GIP secretion is normal or near-normal, whereas GLP-1 secretion is significantly impaired. Potential therapeutic use of GLP-1 for type 2 diabetes was derived from the study demonstrating the glucose-lowering actions of GLP-1 are preserved in patients with type 2 diabetes. To compare insulinotropic actions of exogenous incretin hormones (GIP and GLP-1), normal and type 2 diabetic patients participated in a study. The effect of GIP in type 2 diabetic patients was significantly lower than in normal subjects, whereas GLP-1 retained the glucose-lowering effects in type 2 diabetic patients. In other words, GLP-1 was capable of restoring insulin responses to glucose in type 2 diabetic patients.
- GLP-1 binds to a G-protein-linked receptor expressed on islet β-cells. Binding of GLP-1 to its receptor stimulates insulin secretion in a glucose-dependent manner [24]. Apart from insulinotropic actions and based on a number of effects, GLP-1 is extremely desirable as type 2 diabetes treatment. Firstly, GLP-1 enhances insulin secretion only when glucose levels are high (>250 mg/dL) [25]. GLP-1 potentiates glucose-induced insulin secretion, but does not have any effect on unstimulated insulin secretion and therefore, is unlikely to cause hypoglycemia. Secondly, GLP-1 stimulates not only insulin gene transcription, but also all steps of insulin biosynthesis. Thus, GLP-1 helps to provide a continuous supply of insulin for secretion. Additionally, GLP-1 upregulates genes for the cellular machinery involved in insulin secretion [26] and has been shown capable of providing new β-cells in subjects with insufficient cells [27]. Thirdly, GLP-1 strongly inhibits glucagon secretion. As a result of the studies of GLP-1 infusion in diabetic patients without any residual β-cell secretory capacity, GLP-1 retains glucose-lowering activity due to strong inhibition of glucagon secretion. Further effects of GLP-1 include inhibition of gastrointestinal motility, especially gastric emptying. The slower rate of gastric emptying is desirable in diabetic patients, because postprandial increase of glucose level is reduced. Taken together, these effects render GLP-1 very promising as a therapeutic agent for type 2 diabetes.
- However, a problem limiting the usefulness of GLP-1 in treatment exists. The major drawback for GLP-1 use as a therapeutic agent is the extremely short half-life due to rapid degradation. The rapid initial degradation is due to the ubiquitously expressed enzyme, dipeptidyl peptidase IV (DPP-IV) [28], which cleaves off the 2 N-terminal amino acid residues. The conversion of intact, biologically active GLP-1 to its metabolites occurs with an apparent half-life of 1 to 1.5 minutes.
- After subcutaneous injection of GLP-1, concentrations peak after 30 to 60 minutes and little remains in the circulation after 1 to 2 hours [29] indicating the majority of exogenous GLP-1 is present in the circulation as the truncated metabolite.
- Intravenous infusions of GLP-1 have been reported to completely normalize blood glucose in type 2 diabetic patients [30]. Many researchers have attempted various other types of administration because continuous intravenous infusion is not suitable for routine administration, and the studies have shown the effectiveness of GLP-1. In order to delay the degradation of GLP-1, the properties of the injectable form have been modified. Possibilities include the preparation of GLP-1 with protamine or zinc, as has been done with insulin [31]. However, the results from using various methods have shown the period before degradation is still too short to obtain therapeutic effects from GLP-1 in diabetic subjects. Therefore, multiple injections are required for therapeutic use of GLP-1. Although many practical problems still remain to be solved, a therapy based on GLP-1 is among the most promising recent approaches for the treatment of type 2 diabetes.
- The use of DPP-IV inhibitors was suggested as a result of extreme degradation of GLP-1 in type 2 diabetic patients. Numerous subsequent studies have indicated administration of DPP-IV inhibitors can be offered to subjects at risk of developing diabetes. However, whether DPP-IV inhibition will have long-term effects is unclear. Since GLP-2, an intestinal growth factor is also a substrate of DPP-IV, DPP-IV inhibition may have an effect on intestinal proliferation [32].
- Over the last decade, gene therapy techniques have been developed for introducing genes into cells that alter the properties of the cells. As the technology for introducing new genes into cells has improved, the disease targets for gene therapy have expanded beyond traditional genetic diseases to chronic diseases such as diabetes. If construction of a GLP-1 plasmid with successful delivery is possible, GLP-1 can be produced endogenously. In this case, frequent injection of GLP-1 would no longer be required. Therefore, developing GLP-1 gene delivery system as treatment of type 2 diabetes could be an attractive approach [33].
- In the author's first study, the effect of GLP-1 gene delivery both in vitro and in vivo using a new plasmid constructed with a modified GLP-1 (7-37) cDNA was evaluated. The cDNA contained a furin cleavage site between the start codon and the GLP-1 coding region. The expression of the GLP-1 gene was driven by a chicken β-actin promoter (pβGLP1). The GLP-1 mRNA level was evaluated by RT-PCR 24 hours after transfection. The in vitro results showed a dose-dependent expression of GLP-1. Coculture assay of the GLP-1 plasmid-transfected cells with isolated rat islet cells demonstrated GLP-1 increased insulin secretion by twofold, compared to controls during a hyperglycemic challenge. A single injection of polyethyleneimine/pβGLP1 complex into ZDF rats resulted in increasing insulin secretion and decreasing blood glucose levels that was maintained for 2 weeks. This GLP-1 gene delivery system may provide an effective and safe treatment modality for type 2 diabetes [33].
- In a second study, the effect of GLP1 gene delivery was evaluated both in vitro and in vivo using a new plasmid constructed with a GLP-1 (7-37) cDNA. The expression of the GLP-1 gene was driven by a SV40 promoter/enhancer. To increase the expression level of GLP-1, nuclear factor (NF) κB binding sites were introduced. The in vitro results showed expression of GLP-1 and in vitro activity of GLP-1, which is a glucose-dependent insulinotropic action. A single systemic administration of polyethyleneimine/pSIGLP1NF κB complex into diet-induced obese (DIO) mice resulted in increasing insulin secretion and decreasing blood glucose levels for a duration longer than 2 weeks [34].
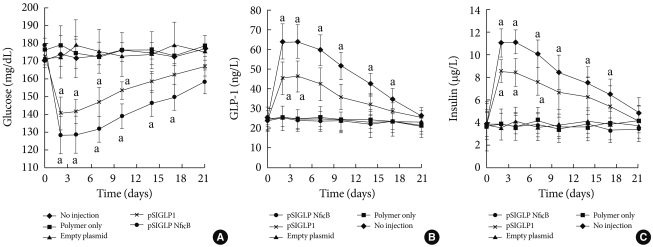
- The expressed GLP-1 should be secreted in its active form for therapeutic purposes. For assessing the active form of GLP-1, ELISA was performed 48 hours after transfection. Fig. 2A shows the amount of GLP-1 in the medium after transfection with each plasmid. Six µg of plasmid DNA complexed with PEI at a N:P ratio of 5:1 was used to transfect the cells in 6-well culture plates. When 6 µg of pSIGLP1N κB-transfected HepG2 cells for GLP-1 production was assayed, the cells produced 1.5-fold more GLP-1 than 6 µg of pSIGLP1-transfected HepG2 cells (130 ng/L/24 hr vs. 201 ng/L/24 hr, P<0.05).
- Whether the secreted GLP-1 from the transfected HepG2 cells can stimulate secretion of insulin from isolated rat islets was also studied. Isolated rat islets were cultured with the pSIGLP-1- and pSIGLP1N κB-transfected HepG2 cells. No enhancement of insulin secretion under low glucose concentrations (50 mg/dL) was observed. However, a significant increment of insulin secretion occurred under high glucose concentrations (300 mg/dL) (Fig. 2B). The transfected HepG2 cells in each well produced 13.2 and 16.3 µg GLP-1 over 4 hours, respectively. The results showed GLP-1 significantly stimulated the secretion of insulin under high glucose conditions but not under low glucose conditions.
- The pSIGLP1/PEI and pSIGLP1N κB/PEI (N:P ratio 5:1) complexes were injected into mice via the tail vein. All mice tolerated the injections well, and no injection-related deaths occurred. Whether the delivery of pSIGLP1N κB plasmid can decrease blood glucose levels in insulin-resistant DIO mice and the effect of incorporating NFB binding sites into the plasmid was studied. After intravenous administration of 200 µg of the PEI/pSIGLP1N κB complex, the blood glucose levels began to decrease (Fig. 3A). The decrease continued until the 2nd day following administration, after which the blood glucose levels increased until the 21st day after injection. However, the blood glucose levels did not return to the preadministration baseline until the 17th day after injection. The control groups showed no significant change in blood glucose levels during a similar time period. The second group of animals that received pSIGLP1/PEI showed a pattern of glucose level change similar to the pSIGLP1N κB/PEI group, but the change of blood glucose in the pSIGLP1 group was relatively smaller than in the pSIGLP1N κB group.
- The plasma level of GLP-1 from each group was also monitored. In conjunction with the changes in blood glucose levels, the PEI/pSIGLP1N κB group plasma GLP-1 concentration began to increase at the 2nd day after injection (Fig. 3B). The GLP-1 level steadily decreased through the 28th day of the study. The control groups showed almost no changes in GLP-1 concentration.
- Plasma insulin levels were assayed to evaluate the insulinotropic effect of GLP-1. The pattern of the insulin concentration change in the PEI/pSIGLP1N κB group mirrored the temporal profile of the plasma GLP-1 levels. The PEI/pSIGLP1N κB group's plasma insulin concentration value increased 2.5-fold above the baseline whereas the control groups showed minimal changes in plasma insulin concentration (Fig. 3C).
- Two days after the complex was injected, plasma GLP-1 values increased dramatically, and the values gradually returned to baseline levels after 3 weeks. Blood glucose levels also decreased and returned to preinjection levels after 3 weeks. Plasma insulin levels also increased and gradually returned to baseline 17 days after injection. An intraperitoneal glucose tolerance test (IPGTT) was performed to verify the improvement of glucose tolerance. The blood glucose level showed a marked decrease.
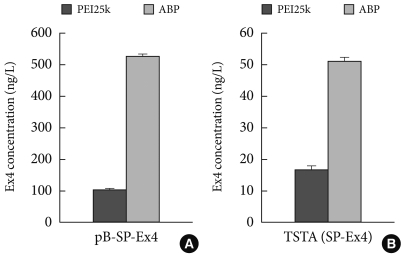
- A third study was performed for gene delivery of exendin-4 (Ex-4). Ex-4 is an analogue of GLP-1, which has resistance to DPP-IV and shows a longer half-life. Therefore, Ex-4 encoding pDNA delivery may be more potent for type 2 diabetes. For gene therapy, pDNA systems for Ex-4 were constructed, which included pβ-SP-Ex4 and TSTA (SP-Ex4) consisting of pβ-Gal-4p65 and pUAS-SP-Ex4. Previously, when PEI25k was used as a gene carrier, a 3:1 mixing ratio between pUAS-SP-Ex4 and pβ-Gal-4p65 reportedly showed the highest Ex-4 level among various ratios in TSTA (SP-Ex4) systems and secreted Ex-4 from that ratio induced the highest insulin level in NIT-1 insulinoma cells. Next, Ex-4 levels from pβ-SP-Ex4 and TSTA (SP-Ex4) were measured and compared after transfections using PEI25k or ABP in NIH3T3 cells. Before performing transfection, the cytotoxicity of PEI25k and araginine-grafted bioreducible polydisulfide amine (ABP) was examined in NIH3T3 cells by MTT assay. As shown in Fig. 4, RCV of PEI25k was abruptly decreased in accordance with the concentration, showing a severe cytotoxicity. However, RCV of ABP was maintained above 100% regardless of the concentration, indicating a minimal cytotoxicity of ABP. Secretion levels of Ex-4 was measured by ELISA in NIH3T3 cells. TATA (SP-Ex-4) pDNA system was prepared at a 3:1 mixing ratio between pUAS-SP-Ex-4 and pβ-Gal4-p65 according to the previous result. The Ex-4 secretion level from TSTA (SP-Ex-4) was found to be approximately 10 times lower than from p β-SP-Ex-4. The result may be due to some type of TSTA (SP-Ex-4) malfunction. However, ABP showed a 3 to 5 fold higher Ex-4 secretion level than PEI25k in both pDNA systems, displaying potency as an efficient Ex-4 gene carrier. Currently, insulin induction activity of secreted Ex-4 is being examined (unpublished data).
GLP-1 GENE DELIVERY
- PCMV-REA-1γ was constructed and characterized. The copolymer of Eph-PEG-TETA.CBA was synthesized and its complex with REA was studied. Mouse islet cells (NIT-1) transfected with PCMV-REA-1γ were treated with isolated NKG2D+CD8+ T-cells. Prevention of the T-cell infiltration into islet by RAE-1γ was investigated by adding NKG2D+CD8+ T-cells to mouse islets. The pancreatic PAE-12 expression profile was observed by immunohistochemistry using anti-PAE-12 NOD mouse. Animal experiments demonstrated the inhibition of type I diabetic progress (unpublished data).
PREVENTION OF TYPE-1 DIABETES USING RAE-1 PLASMID AND EPHRIN CONJUGATED POLYETHYLENE GLYCOL TRIETHYLENE TETRA AMINE/CYSTAMINE BISABRYLAMIDE (EPH-PEG-TETA/CBA) COPOLYMER
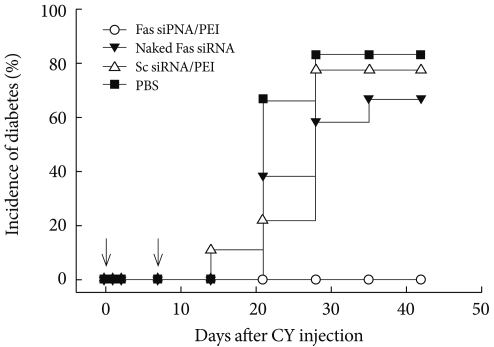
- A membrane receptor, Fas (CD95), and its ligand FasL have been considered as key players in diabetes pathogenesis and are known to mediate interactions between β-cells and cytotoxic T-cells, resulting in apoptotic cell death. The author of the present study hypothesized the interruption of Fas-FasL interactions by suppressing Fas expression in β-cells would affect the development of diabetes. The effect of Fas-silencing siRNA (Fas siRNA) on diabetes development was evaluated in a cyclophosphamide (CY)-accelerated diabetes animal model after intravenous administration using the carrier PEI. The systemic non-viral delivery of Fas siRNA showed significant delay in diabetes incidence up to 40 days, while the control mice treated with naked Fas siRNA, scrambled dsRNA, or PBS were afflicted with diabetes within 20 days. The retardation of diabetes incidence after the treatment of Fas siRNA may be due to the delayed progression of the pancreatic insulitis. In the study, the potential use of a non-viral carrier-based siRNA gene therapy for the prevention of type 1 diabetes is demonstrated [35]. The prevention and effect of Fas siRNA/PEI complexes on CY-induced diabetes in NOD mice are shown in Fig. 5.
Fas siRNA DELIVERY IN CYCLOPHOSPHAMIDE INDUCED WITH DIABETES IN NOD MICE
-
Acknowledgements
- The author wishes to thank K. S. Ko, J. J. Ko, S. Oh, M. Lee, T. Kim, S. Choi, and J. Jeong. Financial supports were by NIH DK077703, DK085075 and WCU 200900000000024.
ACKNOWLEDGMENTS
- 1. Rolland AP. From genes to gene medicines: recent advances in nonviral gene delivery. Crit Rev Ther Drug Carrier Syst 1998;15:143-198. ArticlePubMed
- 2. Peel D. Virus vectors and gene therapy: problems, promises and prospects. MBChB Special Study Module Project Report. 1998. Leicester: Department of Microbiology and Immunology, University of Leicester.
- 3. Anchordoquy TJ, Koe GS. Physical stability of nonviral plasmid-based therapeutics. J Pharm Sci 2000;89:289-296. ArticlePubMed
- 4. Garnett MC. Gene-delivery systems using cationic polymers. Crit Rev Ther Drug Carrier Syst 1999;16:147-207. ArticlePubMed
- 5. Huang L, Hung M, Wagner E. Nonviral vectors for gene therapy. 1999. San Diego: Academic Press; p. 3-22.
- 6. Kawabata K, Takakura Y, Hashida M. The fate of plasmid DNA after intravenous injection in mice: involvement of scavenger receptors in its hepatic uptake. Pharm Res 1995;12:825-830. PubMed
- 7. American Diabetes Association cited 2011 Jan 12. Available from: http://www.diabetes.org.
- 8. Anderson R, Jones P. What's new in type 2 diabetes?. 2000. Liverpool: National Prescribing Centre.
- 9. Lorenzana GG, Hsia SH. The pharmacotherapy of type 2 diabetes to achieve tight glycemic control. Home Health Care Consult 2002;9:10-16.
- 10. Malaisse WJ, Lebrun P. Mechanisms of sulfonylurea-induced insulin release. Diabetes Care 1990;13(Suppl 3):9-17.
- 11. Davies MJ. Insulin secretagogues. Curr Med Res Opin 2002;18(Suppl 1):s22-s30. ArticlePubMed
- 12. Moller DE. New drug targets for type 2 diabetes and the metabolic syndrome. Nature 2001;414:821-827. ArticlePubMedPDF
- 13. Koh JJ, Ko KS, Lee M, Han S, Park JS, Kim SW. Degradable polymeric carrier for the delivery of IL-10 plasmid DNA to prevent autoimmune insulitis of NOD mice. Gene Ther 2000;7:2099-2104. ArticlePubMedPDF
- 14. Lee M, Han S, Ko KS, Kim SW. Cell type specific and glucose responsive expression of interleukin-4 by using insulin promoter and water soluble lipopolymer. J Control Release 2001;75:421-429. ArticlePubMed
- 15. Ko KS, Lee M, Koh JJ, Kim SW. Combined administration of plasmids encoding IL-4 and IL-10 prevents the development of autoimmune diabetes in nonobese diabetic mice. Mol Ther 2001;4:313-316. ArticlePubMed
- 16. Lee M, Han SO, Ko KS, Koh JJ, Park JS, Yoon JW, Kim SW. Repression of GAD autoantigen expression in pancreas beta-Cells by delivery of antisense plasmid/PEG-g-PLL complex. Mol Ther 2001;4:339-346. PubMed
- 17. Jeong JH, Lee M, Kim WJ, Yockman JW, Park TG, Kim YH, Kim SW. Anti-GAD antibody targeted non-viral gene delivery to islet beta cells. J Control Release 2005;107:562-570. ArticlePubMed
- 18. La Barre J, Still EU. Studies on physiology of secretin. III. Further studies on the effects of secretin on blood sugar. Am J Physiol 1930;91:649-653.
- 19. Creutzfeldt W, Ebert R. New developments in the incretin concept. Diabetologia 1985;28:565-573. ArticlePubMedPDF
- 20. Creutzfeldt W. The incretin concept today. Diabetologia 1979;16:75-85. ArticlePubMedPDF
- 21. Beck B. Gastric inhibitory polypeptide: a gut hormone with anabolic functions. J Mol Endocrinol 1989;2:169-174. ArticlePubMed
- 22. Lund PK, Goodman RH, Montminy MR, Dee PC, Habener JF. Anglerfish islet pre-roglucagon II. Nucleotide and corresponding amino acid sequence of the cDNA. J Biol Chem 1983;258:3280-3284. PubMed
- 23. Lopez LC, Frazier ML, Su CJ, Kumar A, Saunders GF. Mammalian pancreatic preproglucagon contains three glucagon-related peptides. Proc Natl Acad Sci U S A 1983;80:5485-5489. ArticlePubMedPMC
- 24. MacDonald PE, El-Kholy W, Riedel MJ, Salapatek AM, Light PE, Wheeler MB. The multiple actions of GLP-1 on the process of glucose-stimulated insulin secretion. Diabetes 2002;51(Suppl 3):S434-S442. ArticlePubMedPDF
- 25. Nathan DM, Schreiber E, Fogel H, Mojsov S, Habener JF. Insulinotropic action of glucagonlike peptide-I-(7-37) in diabetic and nondiabetic subjects. Diabetes Care 1992;15:270-276. ArticlePubMedPDF
- 26. Buteau J, Roduit R, Susini S, Prentki M. Glucagon-like peptide-1 promotes DNA synthesis, activates phosphatidylinositol 3-kinase and increases transcription factor pancreatic and duodenal homeobox gene 1 (PDX-1) DNA binding activity in beta (INS-1)-cells. Diabetologia 1999;42:856-864. ArticlePubMedPDF
- 27. Perfetti R, Zhou J, Doyle ME, Egan JM. Glucagon-like peptide-1 induces cell proliferation and pancreatic-duodenum homeobox-1 expression and increases endocrine cell mass in the pancreas of old, glucose-intolerant rats. Endocrinology 2000;141:4600-4605. ArticlePubMedPDF
- 28. Deacon CF, Johnsen AH, Holst JJ. Degradation of glucagon-like peptide-1 by human plasma in vitro yields an N-terminally truncated peptide that is a major endogenous metabolite in vivo. J Clin Endocrinol Metab 1995;80:952-957. ArticlePubMed
- 29. Drucker DJ. Minireview: the glucagon-like peptides. Endocrinology 2001;142:521-527. ArticlePubMed
- 30. Nauck MA, Kleine N, Orskov C, Holst JJ, Willms B, Creutzfeldt W. Normalization of fasting hyperglycaemia by exogenous glucagon-like peptide 1 (7-36 amide) in type 2 (non-insulin-dependent) diabetic patients. Diabetologia 1993;36:741-744. ArticlePubMedPDF
- 31. Pridal L, Agerbaek H, Christensen LN, Thomsen K, Kirk O. Absorption of glucagons-like peptide-1 can be protracted by zinc or protamine. Int J Pharm 1996;136:53-59.
- 32. Holst JJ, Deacon CF. Inhibition of the activity of dipeptidyl-peptidase IV as a treatment for type 2 diabetes. Diabetes 1998;47:1663-1670. ArticlePubMedPDF
- 33. Oh S, Lee M, Ko KS, Choi S, Kim SW. GLP-1 gene delivery for the treatment of type 2 diabetes. Mol Ther 2003;7:478-483. ArticlePubMed
- 34. Choi S, Oh S, Lee M, Kim SW. Glucagon-like peptide-1 plasmid construction and delivery for the treatment of type 2 diabetes. Mol Ther 2005;12:885-891. ArticlePubMed
- 35. Jeong JH, Kim SH, Lee M, Kim WJ, Park TG, Ko KS, Kim SW. Non-viral systemic delivery of Fas siRNA suppresses cyclophosphamide-induced diabetes in NOD mice. J Control Release 2010;143:88-94. ArticlePubMedPMC
REFERENCES
Fig. 1Suppression of insulitis in NOD mice after tail vein injections of PAGA/DNA complexes (2/1, +/-) at the dose of 100 µg pCAGGS mIL-10 plasmid per mouse. Insulitis was evaluated by hematoxylin-eosin staining of more than 20 islets from each pancreas and evaluating the progression of insulitis. (A) PAGA injected group. (B) PAGA/DNA complex injected group. (C) Naked DNA injected group. (D) Insulitis grade 0. (E) Insulitis grade 2. (F) Insulitis grade 4.

Fig. 2
In vitro transfection assay in HepG2 cells for GLP-1 expression. (A) The GLP-1 levels after transfection of the PEI/pSIGLP1 complex into HepG2 cells. (B) Insulin production in isolated rat islets cocultured with pSIGLP1-transfected HepG2 cells. The graph represents the SE averages for 6 experiments. aP<0.05 compared to control, bP<0.05 compared to pSIGLP1.
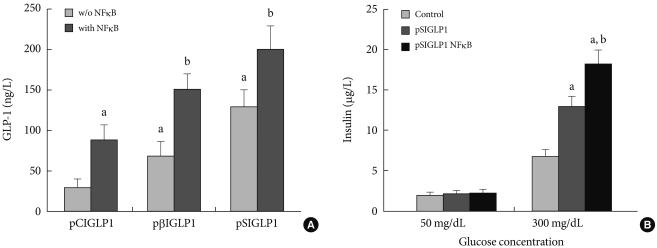
Fig. 3Delivery of PEI/pSIGLP1 in DIO mice. (A) Blood glucose level, (B) plasma GLP-1 level, and (C) plasma insulin concentration changes after PEI/pSIGLP1 injection. The DIO mice received intravenous injection of PEI only or PEI with empty plasmid or PEI with pSIGLP1 or PEI/pSIGLP1. Each group was composed of 6 rats, and the graphs represent the SE averages. aP<0.05 compared to control.
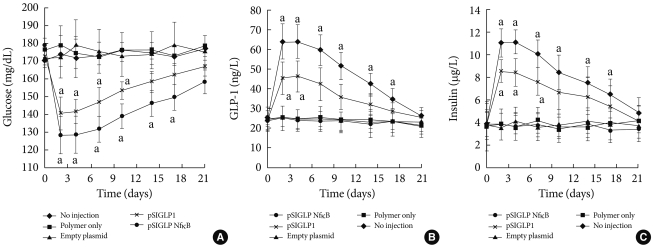


Figure & Data
References
Citations
Citations to this article as recorded by 
- Delivery of therapeutic agents and cells to pancreatic islets: Towards a new era in the treatment of diabetes
Elnaz Zeynaloo, Logan D. Stone, Emre Dikici, Camillo Ricordi, Sapna K. Deo, Leonidas G. Bachas, Sylvia Daunert, Giacomo Lanzoni
Molecular Aspects of Medicine.2022; 83: 101063. CrossRef - Applying emerging technologies to improve diabetes treatment
Yu Jiaojiao, Caifeng Sun, Yuli Wei, Chaoying Wang, Brijesh Dave, Fei Cao, Hu Liandong
Biomedicine & Pharmacotherapy.2018; 108: 1225. CrossRef - Supercritical Fluid-Assisted Decoration of Nanoparticles on Porous Microcontainers for Codelivery of Therapeutics and Inhalation Therapy of Diabetes
Ranjith Kumar Kankala, Xiao-Fen Lin, Hu-Fan Song, Shi-Bin Wang, Da-Yun Yang, Yu Shrike Zhang, Ai-Zheng Chen
ACS Biomaterials Science & Engineering.2018; 4(12): 4225. CrossRef - Glucose‐Responsive Supramolecular Vesicles Based on Water‐Soluble Pillar[5]arene and Pyridylboronic Acid Derivatives for Controlled Insulin Delivery
Lei Gao, Tingting Wang, Keke Jia, Xuan Wu, Chenhao Yao, Wei Shao, Dongmei Zhang, Xiao‐Yu Hu, Leyong Wang
Chemistry – A European Journal.2017; 23(27): 6605. CrossRef - Simultaneous expression and transportation of insulin by supramolecular polysaccharide nanocluster
Yu-Hui Zhang, Ying-Ming Zhang, Qi-Hui Zhao, Yu Liu
Scientific Reports.2016;[Epub] CrossRef - Bioreducible polymers for therapeutic gene delivery
Young Sook Lee, Sung Wan Kim
Journal of Controlled Release.2014; 190: 424. CrossRef - Double-strand adeno-associated virus-mediated exendin-4 expression in salivary glands is efficient in a diabetic rat model
Junhong Wang, Feng Wang, Jing Xu, Shimei Ding, Yonghong Guo
Diabetes Research and Clinical Practice.2014; 103(3): 466. CrossRef - Polymer-Based Delivery of Glucagon-Like Peptide-1 for the Treatment of Diabetes
Pyung-Hwan Kim, Sung Wan Kim
ISRN ENDOCRINOLOGY.2012; 2012: 1. CrossRef - High serum ferritin levels are associated with metabolic risk factors in non‐obese Korean young adults: Korean National Health and Nutrition Examination Survey (KNHANES) IV
Ki‐Dong Yoo, Seung‐Hyun Ko, Ji‐Eun Park, Yu‐Bae Ahn, Hyeon Woo Yim, Won‐Chul Lee, Yong‐Moon Park
Clinical Endocrinology.2012; 77(2): 233. CrossRef - Delivery of two-step transcription amplification exendin-4 plasmid system with arginine-grafted bioreducible polymer in type 2 diabetes animal model
Pyung-Hwan Kim, Minhyung Lee, Sung Wan Kim
Journal of Controlled Release.2012; 162(1): 9. CrossRef - Polymeric delivery of therapeutic RAE-1 plasmid to the pancreatic islets for the prevention of type 1 diabetes
Wan Seok Joo, Ji Hoon Jeong, Kihoon Nam, Katherine S. Blevins, Mohamed E. Salama, Sung Wan Kim
Journal of Controlled Release.2012; 162(3): 606. CrossRef
 PubReader
PubReader Cite
Cite