- Current
- Browse
- Collections
-
For contributors
- For Authors
- Instructions to authors
- Article processing charge
- e-submission
- For Reviewers
- Instructions for reviewers
- How to become a reviewer
- Best reviewers
- For Readers
- Readership
- Subscription
- Permission guidelines
- About
- Editorial policy
Articles
- Page Path
- HOME > Diabetes Metab J > Volume 47(3); 2023 > Article
-
Original ArticleBasic Research Hyperglycemia-Suppressed SMARCA5 Disrupts Transcriptional Homeostasis to Facilitate Endothelial Dysfunction in Diabetes
-
Ju Wang1
 , Hui Zhou2, Jinhua Shao2, Shu Zhang2, Jing Jin2
, Hui Zhou2, Jinhua Shao2, Shu Zhang2, Jing Jin2 -
Diabetes & Metabolism Journal 2023;47(3):366-381.
DOI: https://doi.org/10.4093/dmj.2022.0179
Published online: March 6, 2023
- 1,662 Views
- 98 Download
1Department of Nursing, Xi’an No. 5 Hospital, Xi’an, China
2Department of Geriatric, Xi’an No. 5 Hospital, Xi’an, China
-
Corresponding author: Jing Jin Department of Geriatric, Xi’an No. 5 Hospital, Xi’an Institute of Rheumatology, No. 112 Xi Guan Zheng Street, Lian Hu District, Xi’an 710082, China E-mail: ljjdoctor@163.com
Copyright © 2023 Korean Diabetes Association
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (https://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
-
Background
- Dysfunction of vascular endothelial cells (ECs) plays a central role in the pathogenesis of cardiovascular complications in diabetes. SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily A member 5 (SMARCA5) is a key regulator of chromatin structure and DNA repair, but its role in ECs remains surprisingly unexplored. The current study was designed to elucidate the regulated expression and function of SMARCA5 in diabetic ECs.
-
Methods
- SMARCA5 expression was evaluated in ECs from diabetic mouse and human circulating CD34+ cells using quantitative reverse transcription polymerase chain reaction and Western blot. Effects of SMARCA5 manipulation on ECs function were evaluated using cell migration, in vitro tube formation and in vivo wound healing assays. Interaction among oxidative stress, SMARCA5 and transcriptional reprogramming was elucidated using luciferase reporter assay, electrophoretic mobility shift assay and chromatin immunoprecipitation.
-
Results
- Endothelial SMARCA5 expression was significantly decreased in diabetic rodents and humans. Hyperglycemia-suppressed SMARCA5 impaired EC migration and tube formation in vitro, and blunted vasculogenesis in vivo. Contrarily, overexpression of SMARCA5 in situ by a SMARCA5 adenovirus-incorporated hydrogel effectively promoted the rate of wound healing in a dorsal skin punch injury model of diabetic mice. Mechanistically, hyperglycemia-elicited oxidative stress suppressed SMARCA5 transactivation in a signal transducer and activator of transcription 3 (STAT3)-dependent manner. Moreover, SMARCA5 maintained transcriptional homeostasis of several pro-angiogenic factors through both direct and indirect chromatin-remodeling mechanisms. In contrast, depletion of SMARCA5 disrupted transcriptional homeostasis to render ECs unresponsive to established angiogenic factors, which ultimately resulted in endothelial dysfunction in diabetes.
-
Conclusion
- Suppression of endothelial SMARCA5 contributes to, at least in part, multiple aspects of endothelial dysfunction, which may thereby exacerbate cardiovascular complications in diabetes.
- As a major public health problem worldwide, diabetes mellitus (DM) exerts a grave influence on the vascular endothelium. The alterations in vascular endothelial cells (ECs) include impaired cell migration, hyporesponsiveness or unresponsiveness to angiogenic stimulation, blunted wound healing and suppressed vessel re-endothelialization [1]. In turn, vascular endothelial dysfunction induced by hyperglycemia is thought to be one of the key risk factors that predispose microvascular diseases, ultimately leading to high morbidity and mortality. To this end, restoration of endothelial function represents a promising strategy for the treatment of diabetic vascular complications [2]. The defective angiogenesis may partially be caused by reduced expression of vascular endothelial growth factor (VEGF) and growth factors. Because compelling evidence points to that VEGF may be beneficial for patients with diabetes and coronary artery or peripheral vascular disease since it may induce the development of cardiac and limb vascular collateralization, respectively [3]. Other biochemical events have been also proposed to underpin diabetic endothelial dysfunction, including increased advance glycation end products and matrix metalloproteinases, sorbitol-inositol imbalance, oxidative stress, increased free fatty acids and cytokines and reduced fibroblast growth factor and endothelial progenitor cell (EPC) circulation [4]. Nevertheless, the exact mechanisms by which angiogenic response is compromised in diabetes remain poorly defined.
- The regulation of noncovalent ordering of chromatin structure by the ATPase-dependent chromatin remodelers, including switch/sucrose nonfermentable (SWI/SNF), imitation SWI, chromodomain helicase DNA binding (CHD), and inositol requiring 80, has become a popular research interest due to their vigorous roles in altering gene expression and modulating cell fate specification [5]. Among the members of the SWI/SNF family, SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily A member 5 (SMARCA5)/chromodomain helicase DNA binding protein 4 (CHD4) has emerged as major integrator of multiple signaling cascades in transformed cells, due to its capacity not only to interplay with cell proliferation and DNA damage repair related signaling [6], but also to sustain hematopoietic cell transformation via interaction with oncofusion proteins [7]. Likewise, deregulated SMARCA5 expression has been functionally linked to leukemia [8], glioblastoma [9] and breast [10], lung [11], and gastric cancers [12]. Nonetheless, information about the potential contribution SMARCA5 in the regulation of the non-malignant actions remains surprisingly limited, despite recent biochemical evidence in a genetically engineered Zebrafish model suggesting that SMARCA5 signaling may regulate erythrocyte aggregation or development of hematopoietic stem and progenitor cells (HSPCs) via its epigenetic programming ability [13,14]. In the present study, we aimed to characterize more deeply the expression pattern and possible roles of SMARCA5 in the potential regulation of EC function. Using a combination of histological, pharmacological and biochemical assays, we present evidence for an important physiological role of SMARCA5 in ECs as a putative modulator of transcriptional homeostasis during “angiogenic switch,” thereby controlling endothelial migration and angiogenesis in vivo. By contrast, repression of SMARCA5 expression by hyperglycemia may mediates, at least in part, vascular dysfunction in diabetes.
INTRODUCTION
- Animal model
- Ten-week-old Balb/c male mice were obtained from the animal facility in Xi’an No. 5 Hospital. Mice were housed in groups of 5, with free access to food pellets and ad libitum water, in a 20°C to 25°C environment. Animals were allowed to acclimatize for at least 1 week before experiments [15]. Type 1 diabetes mellitus (T1DM) was induced via intraperitoneal administration of a single dose of streptozotocin (STZ, 160 mg/kg/body weight, dissolved in 200 μL of 10 mM sodium citrate buffer, pH 4.0, Sigma-Aldrich, Shanghai, China), as described elsewhere [16]. The mice with blood glucose ≥250 mg/dL 2 weeks following injection were considered diabetic [17]. To induce type 2 diabetes mellitus (T2DM), were fed ad libitum a high-fat diet (HFD) containing 60 kcal% fat (TROPHIC Animal Feed High-tech, Nantong, China) for 10 weeks. Eight weeks following HFD feeding, mice were injected intraperitoneally with one dose of STZ (100 mg/kg/body weight) [18]. Mice with distinct hyperglycemia concomitant with insulin resistance (beyond 1 to 2 standard error of the mean values from the normal mice mean), at 3 weeks after STZ injection, were considered diabetic [17]. All procedures involved in animal work were strictly conformed to the guidelines of the Institutional Animal Care and Use by National Institutes of Health, and was approved by the Institutional Animal Care and Use Committee of Xi’an No. 5 Hospital (Approval #: XAFH-2016-0081a).
- Detailed information on other experimental procedures and statistical analysis pertinent to this paper can be found in Supplementary Methods and Supplementary Table 1.
METHODS
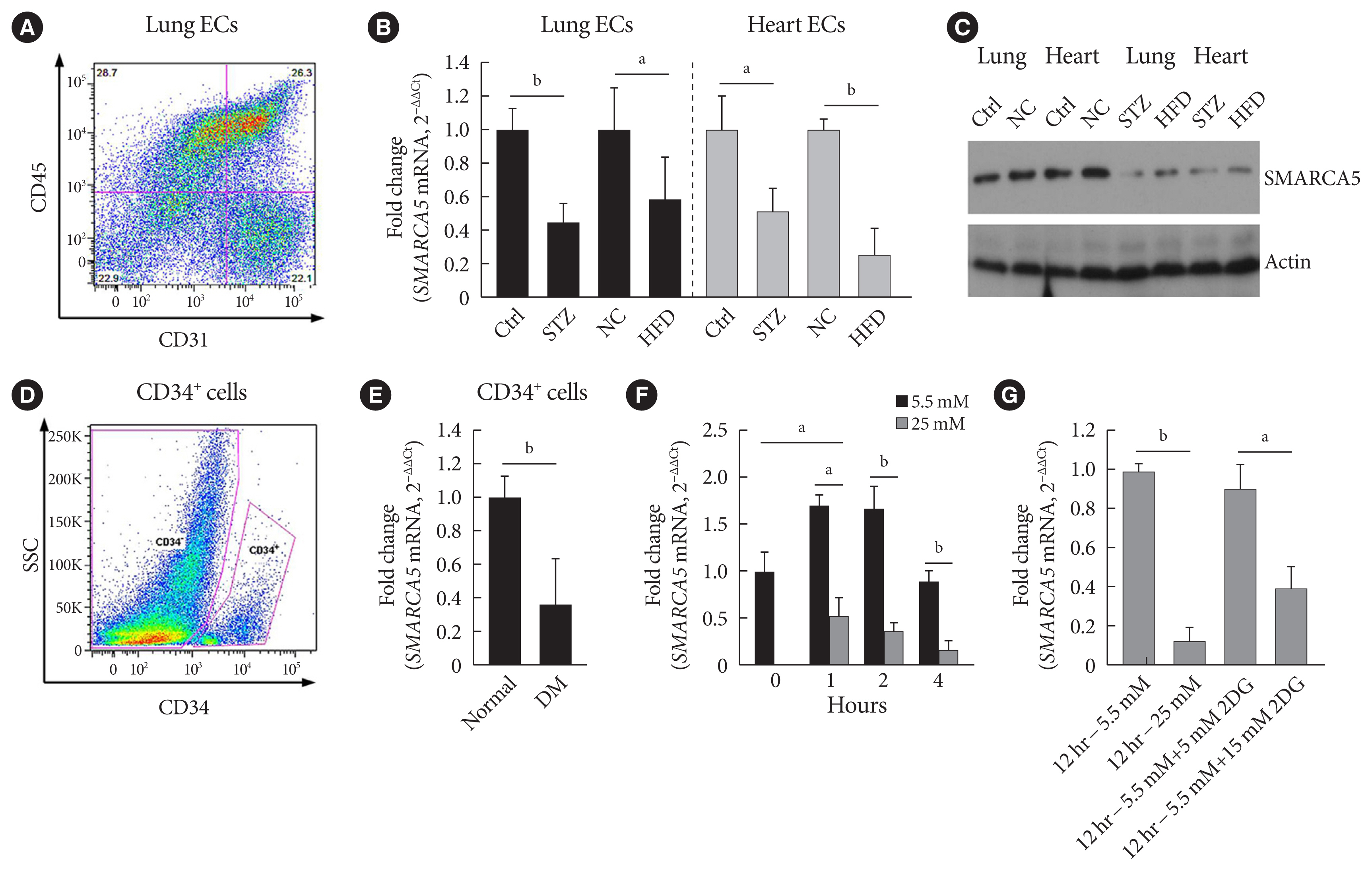
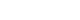
- Diabetes impairs SMARCA5 expression in ECs
- Because we were interested in the potential involvement of SMARCA5 in the regulation of diabetes-induced endothelial dysfunction, we initially investigated the effects of diabetes on Smarca5 expression in ECs in vivo (Fig. 1A). As revealed by quantitative polymerase chain reaction (qPCR), Smarca5 expression was reduced by more than 30% in ECs isolated from either lung or heart tissues of these diabetic mice, compared to control mice (Fig. 1B). The deleterious effects of diabetes on SMARCA5 expression in ECs was further validated at protein level using Western blot (Fig. 1C). Similarly, qPCR analysis in human circulating CD34+ cells (Fig. 1D), which consist of significant numbers of EPCs [19,20], also demonstrated significant reduced expression levels of SMARCA5 in patients with T2DM, as compared to control subjects (0.36±0.27 vs. 1.00± 0.15, P<0.01) (Fig. 1E). We conducted this assay to determine expression difference of SMARCA5 in circulating CD34+ cells from patients with T2DM, based on evaluation of mean HbA1c (5.4±0.3 in normal vs. 8.2±0.2 in DM, P<0.0001) (Supplementary Fig. 1). The observation that both T2DM and T1DM impaired SMARCA5 expression in ECs pointed to the possibility that hyperglycemia, rather than hyperinsulinemia, may negatively regulate endothelial SMARCA5 expression [21]. To test this, primary lung ECs were cultured in high glucose (25 mM) or placed in physiological glucose concentrations (5.5 mM) [20]. Expression levels of Smarca5 rapidly declined by 70% within 2 hours and by 85% within 4 hours in high glucose, respectively (Fig. 1F). Conversely, this inhibitory effect of hyperglycemia was substantially reversed by the addition of 2-deoxy glucose, a competitive inhibitor of glucose metabolism (Fig. 1G) [22]. Thus, diabetes, likely at least in part via hyperglycemia, attenuates SMARCA5 expression in ECs, and likely in humans as well.
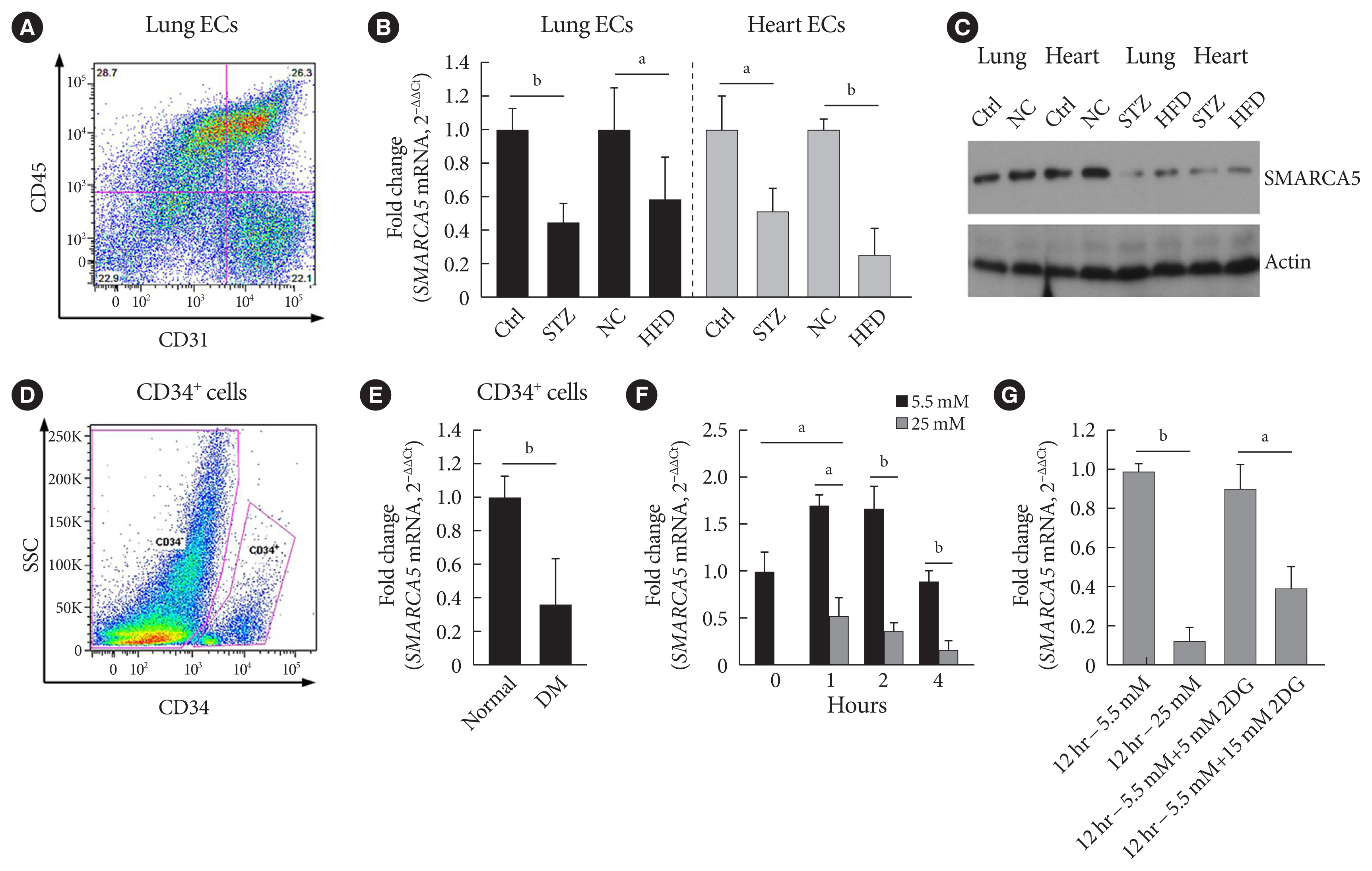
- SMARCA5 deficiency compromises EC migration and tube formation
- EC migration is essential for angiogenesis and is usually blunted in diabetes [23]. We next verified the effects of intact SMARCA5 expression on EC migration in vitro. Stable knockdown of SMARCA5 was induced in human umbilical vein endothelial cells (HUVECs) by short hairpin RNA (shRNA) lentivirus infection, and the knockdown efficiency of shRNA targeting SMARCA5 was verified by qPCR (Fig. 2A) and Western blot (Fig. 2B) at mRNA and protein levels, respectively. HUVECsSMARCA5−/− cells displayed about a 45.4% and 45.0% reduction in their relative migration ability at 24 hours following generation of scratch migration assay respectively, as compared to that in mock control and scramble shRNA-treated HUVECs (Fig. 2C). Similarly, during sphingosine 1-phosphate (S1P)-induced angiogenesis, HUVECsSMARCA5−/− also exhibited significantly repressed migration compared to mock control and scramble shRNA-treated HUVECs, as measured by transwell migration assays (Fig. 2D). Ablation of SMARCA5 was also inhibited the formation ability of tube-like structures by approximately 65.0% in HUVECs (Fig. 2E). Thus, SMARCA5 deficiency sabotages endothelial function under both resting and stimulated conditions. S1P acts through distinct cell surface receptors that involves at least two intracellular signaling cascades: the extracellular regulated protein kinase (ERK) and Akt/endothelial nitric oxide synthase (eNOS) pathways [24]. To further dissect the molecular basis underpinning our observation, HUVECs with different transfections were stimulated with S1P, and activation of Akt, eNOS, and ERK was measured by phospho-protein Western blot. Apparently, SMARCA5 depletion had profound impact on activation of both ERK and Akt pathways in S1P-stimulated HUVECs (Fig. 2F). Together, our findings suggest that endothelial SMARCA5 is a positive regulator of angiogenic activation.
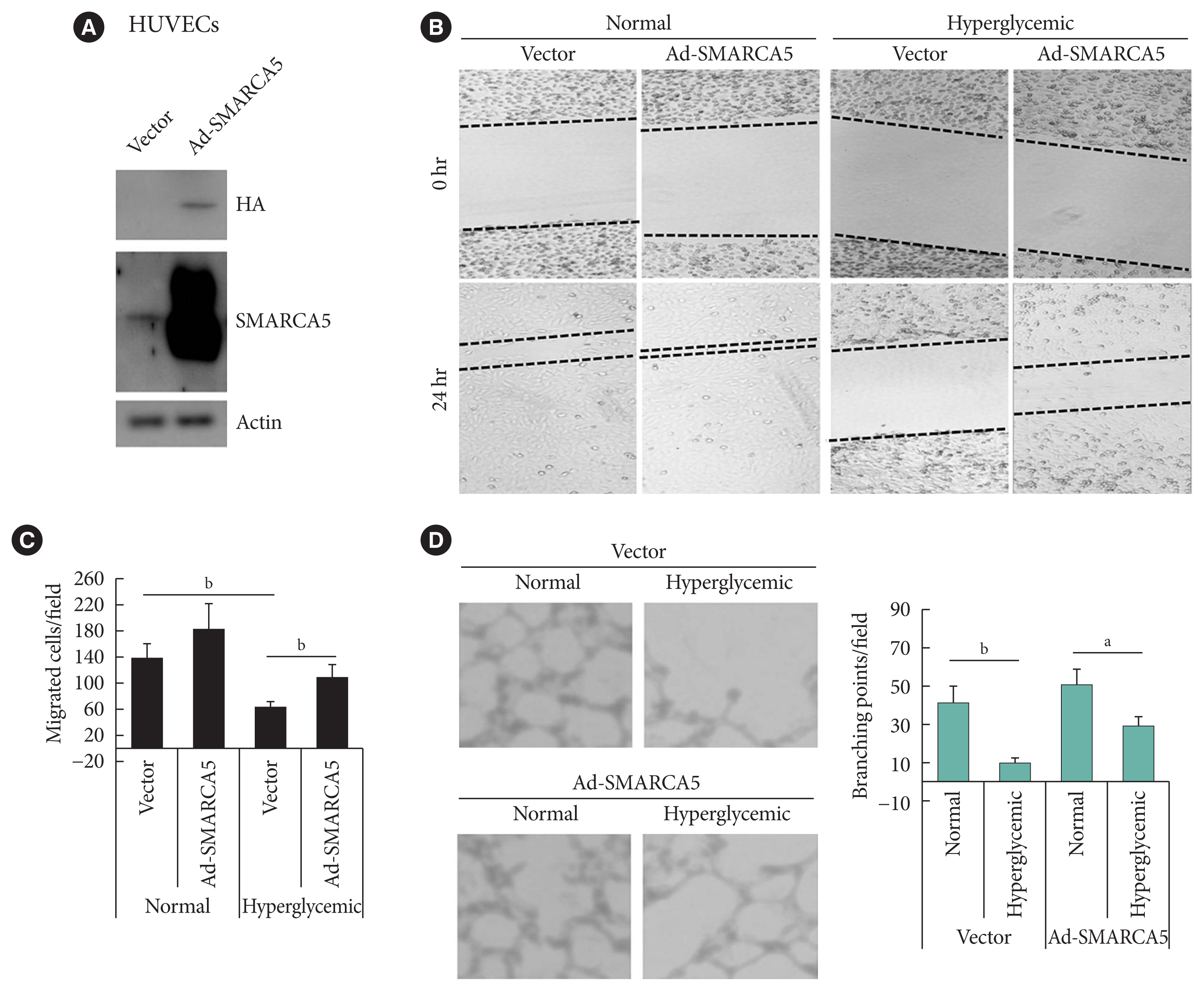
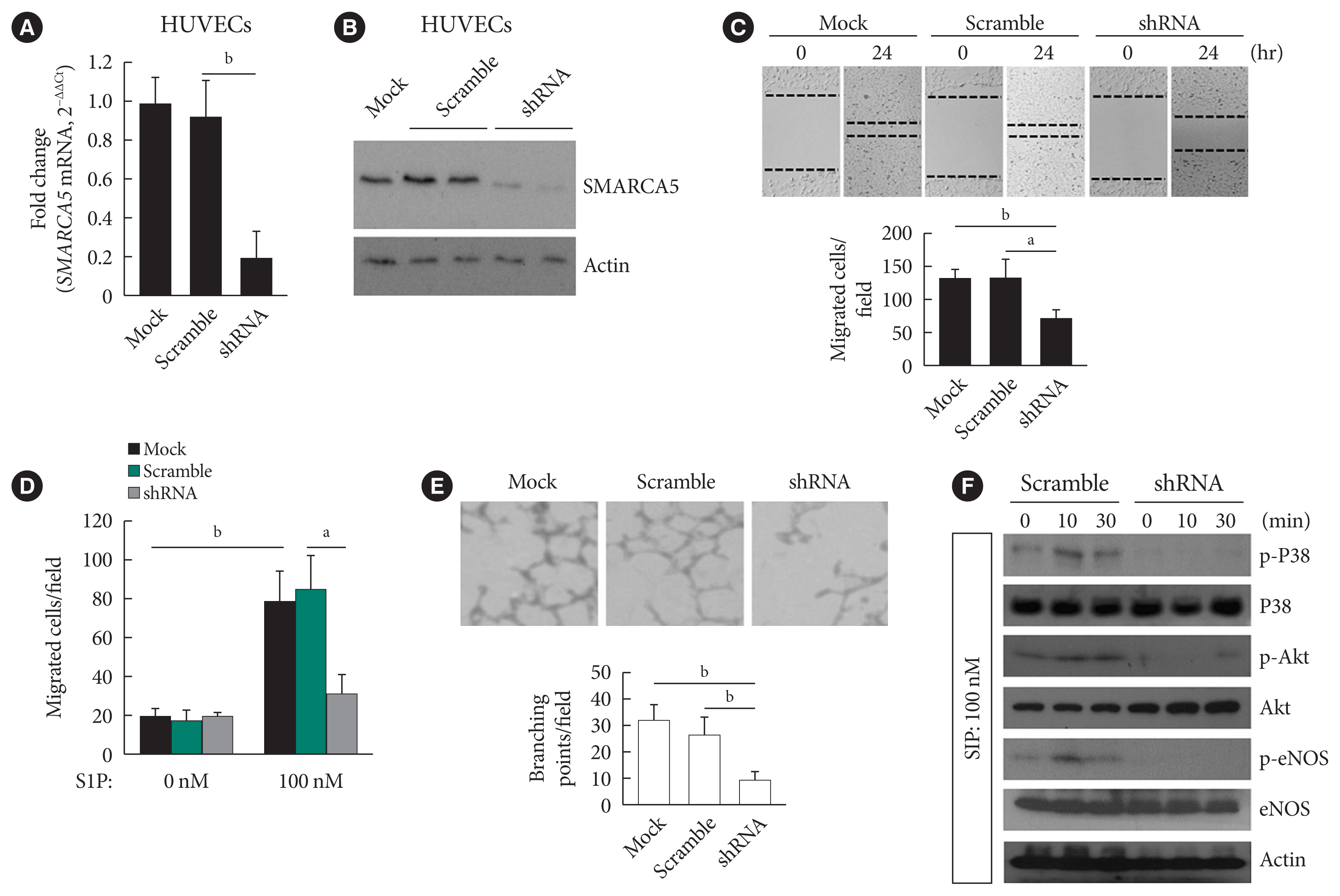
- SMARCA5 overexpression ameliorates EC migration and tube formation under hyperglycemic conditions
- To ask directly whether SMARCA5 regulates EC functions, as inferred from the above-mentioned loss-of-function analysis, we generated the HUVECs that stably overexpressed the exogenous SMARCA5 (HUVECs/Ad-SMARCA5) (Fig. 3A), and then cultured the cell under hyperglycemic conditions versus normal glycemic conditions. Hyperglycemia-suppressed endothelial migration was significantly attenuated in HUVECs/Ad-SMARCA5 cells compared with the vector-transfected cells (110.8±19.8 in Ad-SMARCA5 plus hyperglycemia vs. 63.8± 10.6 in vector plus hyperglycemia, P<0.01) (Fig. 3B and C). In agreement, tube formation rate was substantially augmented in HUVECs cells by SMARCA5 overexpression (Fig. 3D). Thus, SMARCA5 directly promotes restoration of EC functions under hyperglycemic conditions.
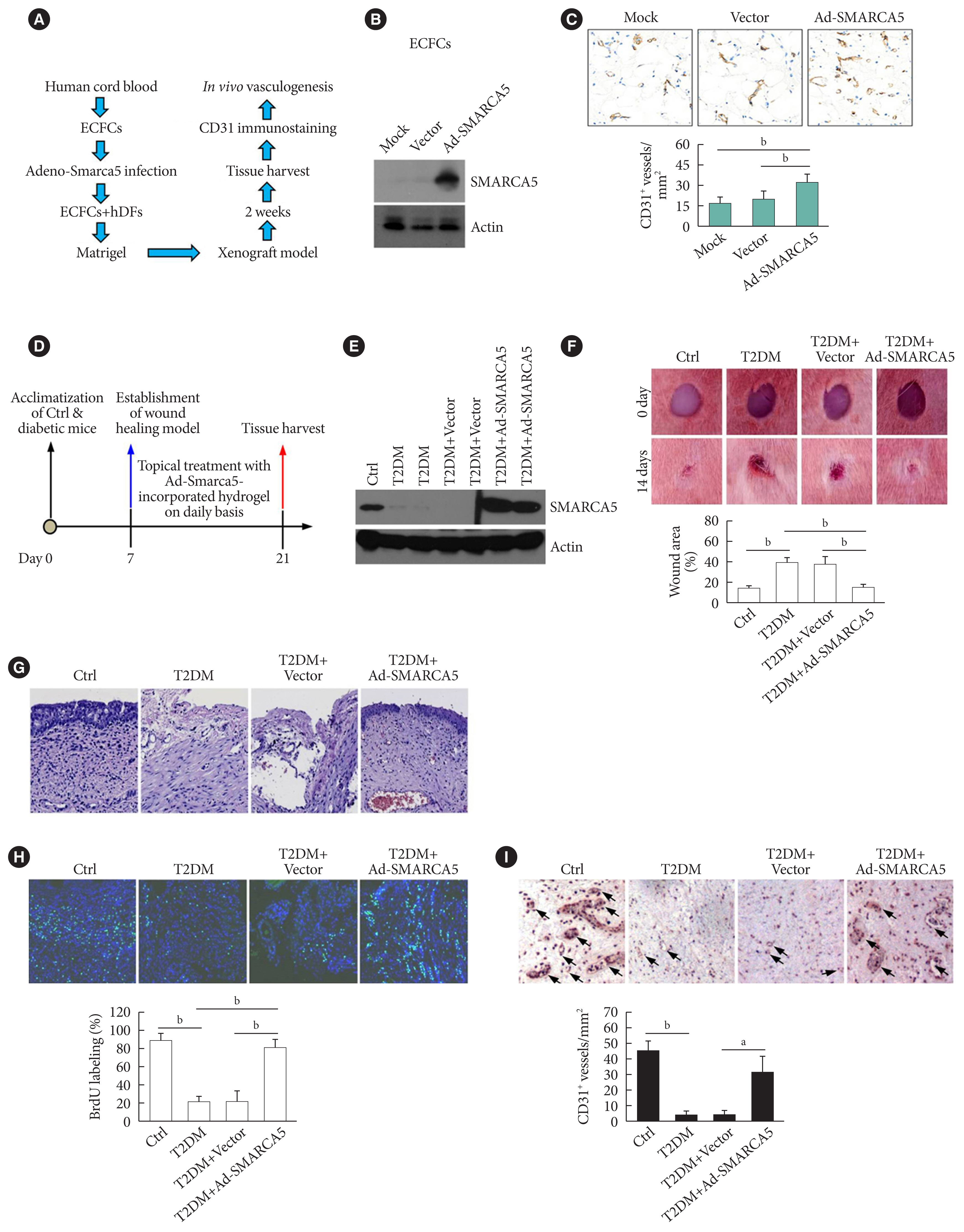
- Replenishment of SMARCA5 potentiates angiogenesis in vivo
- Because EC migration is indispensible for the formation of blood vessels [20], we were curious whether SMARCA5 is functionally involved in angiogenesis in vivo. We transfected endothelial colony forming cells (ECFCs) with SMARCA5 adenovirus and then co-injected these ECFCs along with human dermal fibroblast (hDFs) subcutaneously onto the dorsal side of nude mice (Fig. 4A). At 2 weeks after cell implantation, ectopic overexpression of SMARCA5 in implants was validated by Western blot (Fig. 4B). Accordingly, immunohistochemical analysis of CD31+ vessels revealed that injection of ECFCs that overexpress SMARCA5 noticeably stimulated the formation of new blood vessels, compared to the use of mock or vector-transfected ECFCs (Fig. 4C). As EC migration is crucial for re-endothelialization of vessel walls following injury, which is dysfunctional in diabetes [20], we next investigated the wound healing effects of the SMARCA5 adenovirus-enriched hydrogel dressings on a skin biopsy punch model (Fig. 4D) [25]. Western blot analysis using subcutaneous tissue homogenates at 14 days after topical treatment with SMARCA5 adenovirus-enriched hydrogel demonstrated a more than three-fold increase in SMARCA5 expression in the wounds that were treated with Ad-SMARCA5 compared to mock or vector-treated wounds (Fig. 4E), thus ensuring the efficiency of SMARCA5 overexpression in vivo. Accordingly, ectopic overexpression of SMARCA5 strongly promoted the rate of wound healing in a dorsal skin punch injury model (16.2±3.1 vs. 38.9±5.9 relative wound area for T2DM+Ad-Smarca5 vs. T2DM+vector, P<0.01) (Fig. 4F). Subsequent histological examination showed that augmentation of SMARCA5 expression in situ significantly promoted reepithelization in healing wounds (Fig. 4G). Considering that reepithelialization is the process of creating a new barrier between wound and environment through epithelial cell proliferation and migration [26], we reasoned that subcutaneous cell proliferation could be enhanced by SMARCA5 overexpression. To test this, 5-bromodeoxyuridine (BrdU) was used to label dividing cells. Quantification of the BrdU-positive cells in subcutaneous tissue revealed an approximately 79.7% reduction in BrdU labeling in T2DM mice, whereas this inhibitory effect of T2DM was effectively abolished by topical treatment with Ad-Smarca5-incorporated hydrogel in the dorsal skin punch injury model (Fig. 4H). In agreement, overexpress SMARCA5 significantly stimulated the formation of subcutaneous new blood vessels, as revealed by immunohistochemical analysis of CD31+ vessels (Fig. 4I). Collectively, the available data indicate that vascular complications of diabetes are mediated, at least in part, via the attenuation of SMARCA5 expression in the endothelium.
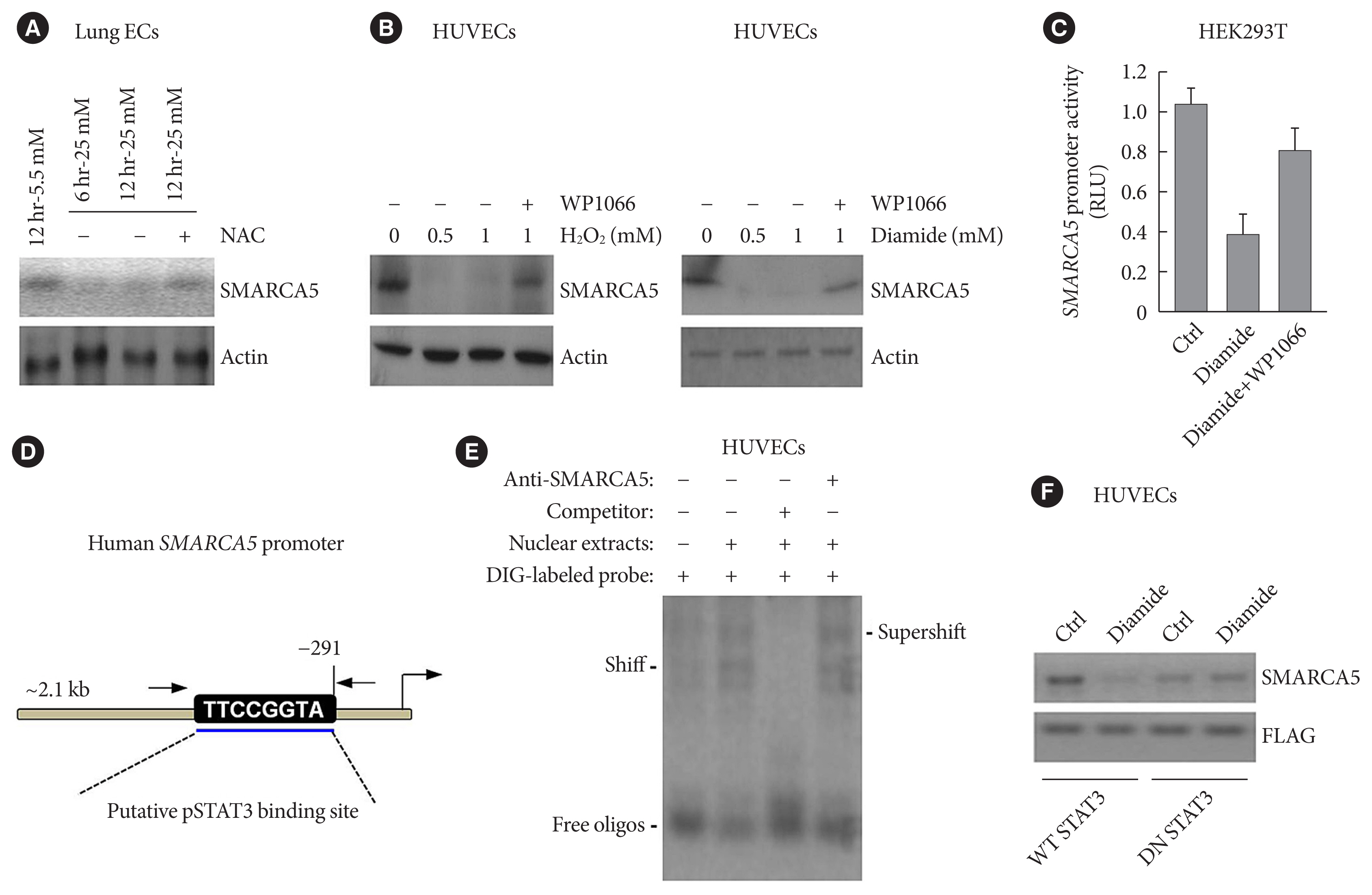
- Hyperglycemia-elicited oxidative stress represses SMARCA5 expression in a STAT3-dependent manner
- Since hyperglycemia-induced oxidative stress is a well-known factor causing endothelial dysfunction that plays a central role in the pathogenesis of cardiovascular complications of diabetes [27], we sought to know whether aberrant oxidative stress is involved in hyperglycemia-induced inhibition of SMARCA5. Lung ECs were incubated with 5.5 or 25 mM glucose for 6 or 12 hours. Treatment with high dose of glucose inhibited SMARCA5 expression in a time-dependent manner, whereas co-treatment with the reactive oxygen species (ROS) scavenger-N-acetyl-L-cysteine (NAC) completely neutralized the inhibitory effects of hyperglycemia on SMARCA5 expression (Fig. 5A). To provide a more persuasive evidence, HUVECs cells were challenged with 0.5 or 1 mM of H2O2 or an oxidative stress inducer diamide for 12 hours [28]. Treatment with both oxidative stress inducers noticeably suppressed SMARCA5 expression, and intriguingly, co-treatment with WP1066 (an inhibitor of signal transducer and activator of transcription 3 [STAT3] activation [29]) significantly neutralized these inhibitory effects (Fig. 5B). We then performed luciferase assay to see whether the inhibition of SMARCA5 expression by diamide was due to a direct transcriptional effect. Incubation with diamide for 12 hours caused an approximately 61.2% reduction in luciferase activity relative to control, while co-treatment with WP1066 effectively restored the luciferase activity in diamide-challenged cells (Fig. 5C). Thus, oxidative stress can directly modulate SMARCA5 expression on a transcriptional level, probably via activation of STAT3 signaling. Indeed, in silico analysis (Ensembl and University of California Santa Cruz [UCSC] databases) of the approximately 2.1 kB of the human SMARCA5 promoter revealed a putative pSTAT3 binding site (Fig. 5D). We next used electrophoretic mobility shift assay to further examine direct binding of pSTAT3 onto the SMARCA5 chromatin. pSTAT3 in the nuclear extracts from diamide-treated SMARCA5/STAT3 HUVECs clearly formed a shifted digoxigenin (DIG)-labeled band, which was supershifted by anti-pSTAT3 antibody. The band was totally abrogated by competition with a 50-fold excess of cold probe (Fig. 5E). To further confirm that functional STAT3 signaling is necessary for oxidative stress’s action to repress SMARCA5, we transduced HUVECs with wild-type STAT3 (WT STAT3) or dominant-negative STAT3 (DN STAT3), and subjected these cells to diamide challenge for 12 hours. Transfection with WT STAT3 caused a robust reduction in SMARCA5 levels but transfection with DN STAT3 failed to attenuate SMARCA5 expression in diamide-challenged HUVECs (Fig. 5F). Together, STAT3 activation is indispensable in oxidative stress’s action to inhibit SMARCA5 expression.
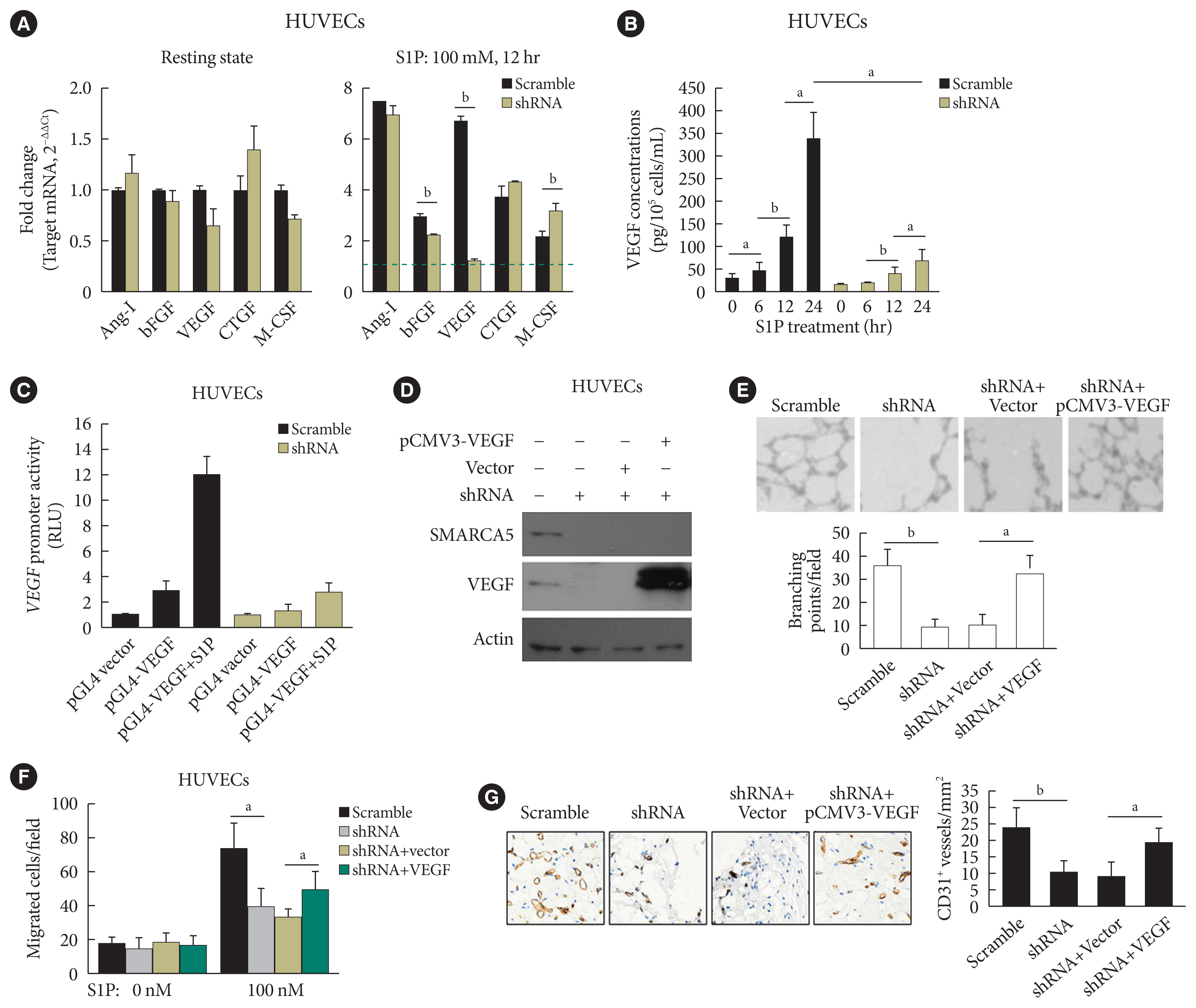
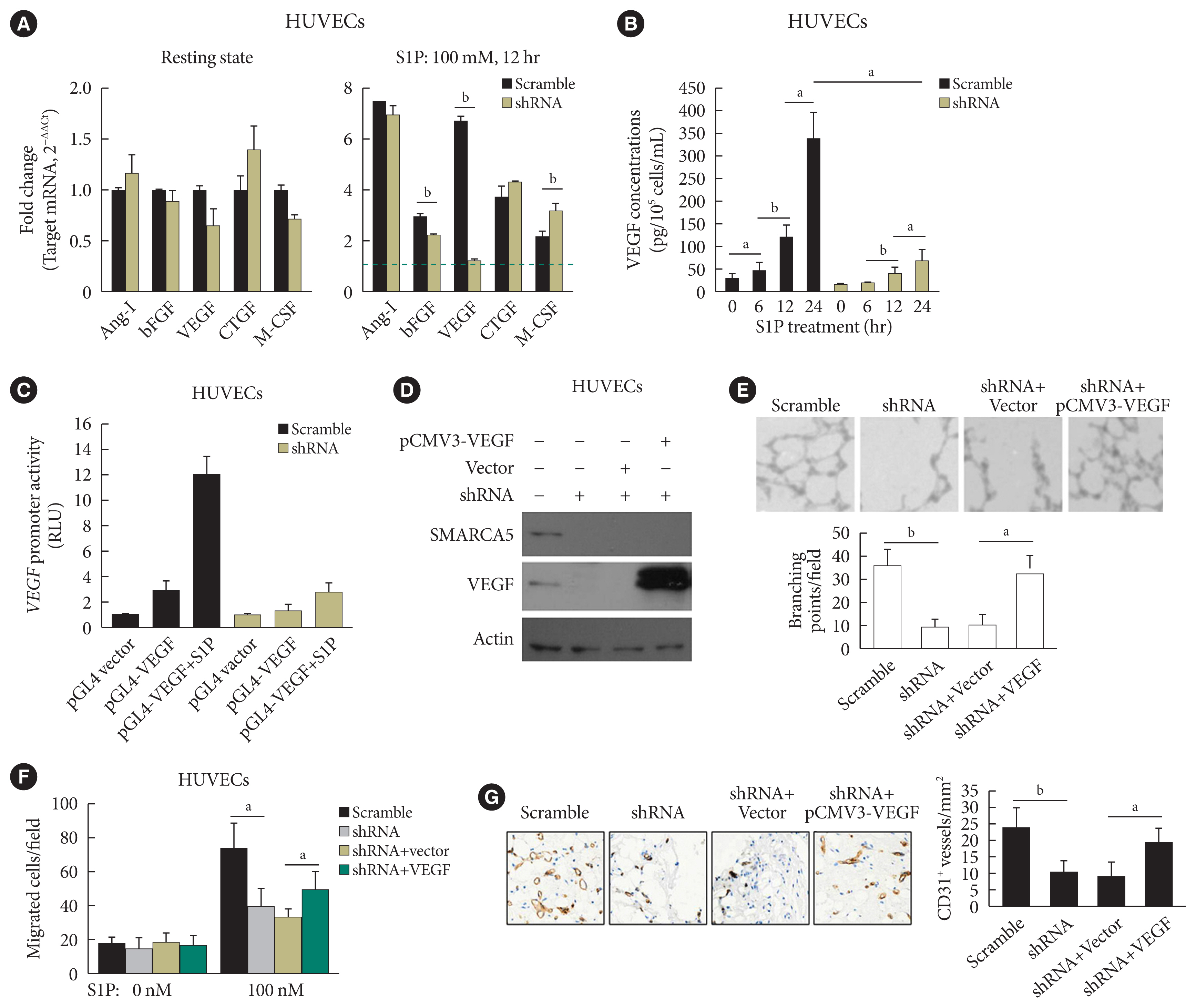
- SMARCA5 activates VEGF signaling to promote endothelial function
- As further exploration of the core signaling pathway responsible for SMARCA5 action during angiogenesis, we evaluated the expression levels of pro-angiogenic factors known to be essential for vascularization [30], in Scramble shRNA or SMARCA5 shRNA-transfected HUVECs. Interestingly, upon S1P challenge, expression levels of basic fibroblast growth factor (bFGF) and VEGF were constantly decreased in SMARCA5 shRNA-transfected HUVECs compared to Scramble shRNA-treated HUVECs, with the lower values being observed in the latter (1.24±0.10 vs. 6.71±0.18 VEGF mRNA expression for SMARCA5 shRNA-transfected HUVECs vs. scramble shRNA-treated HUVECs, P<0.01). By contrast, SMARCA5 depletion had no effects on expression levels of these pro-angiogenic factors under resting state (Fig. 6A). Consistently, confirmation of attenuation of VEGF expression upon SMARCA5 deficiency, at protein level, was achieved by measuring VEGF concentration in the supernatants using an ELISA assay, which revealed that the time-dependent induction of VEGF in S1P-stimulated HUVECs was substantially compromised by SMARCA5 shRNA treatment (68.67±26.65 vs. 339.60±57.52 VEGF concentration at 24 hours for SMARCA5 shRNA-transfected HUVECs vs. scramble shRNA-treated HUVECs, P<0.05) (Fig. 6B). To investigate the transcriptional activity of the SMARCA5 gene in different cell types, pGL4.10-VEGF was transfected into scramble shRNA or SMARCA5 shRNA-transfected HUVECs for 48 hours, followed by treatment with S1P for another 6 hours. SMARCA5 shRNA-transfected HUVECs exhibited the lower promoter activity (2.73±0.82 renilla luciferase activity [RLU]) when compared to scramble shRNA-treated HUVECs (12.07±1.34 RLU) (Fig. 6C), indicating an association between SMARCA5 depletion and dysfunction in VEGF synthesis in ECs. To address the possibility that SMARCA5 may exert its regulatory role through a direct modulation of the VEGF signaling in stimulated ECs, we stably overexpressed the exogenous VEGF in HUVECsSMARCA5−/− cells. Notably, overexpression of VEGF failed to restore the expression levels of SMARCA5 in HUVECsSMARCA5−/− cells, reemphasizing the notion that SMARCA5 may act downstream of the VEGF signaling (Fig. 6D). Consequently, SMARCA5 depletion-induced impaired tube formation (Fig. 6E), blunted cell migration (Fig. 6F), as well as suppressed vessel reendothelialization in vivo (Fig. 6G), were all effectively ameliorated by VEGF overexpression in HUVECsSMARCA5−/− cells. These findings unequivocally suggest that VEGF may function as a major downstream effector of SMARCA5 in stimulated ECs.
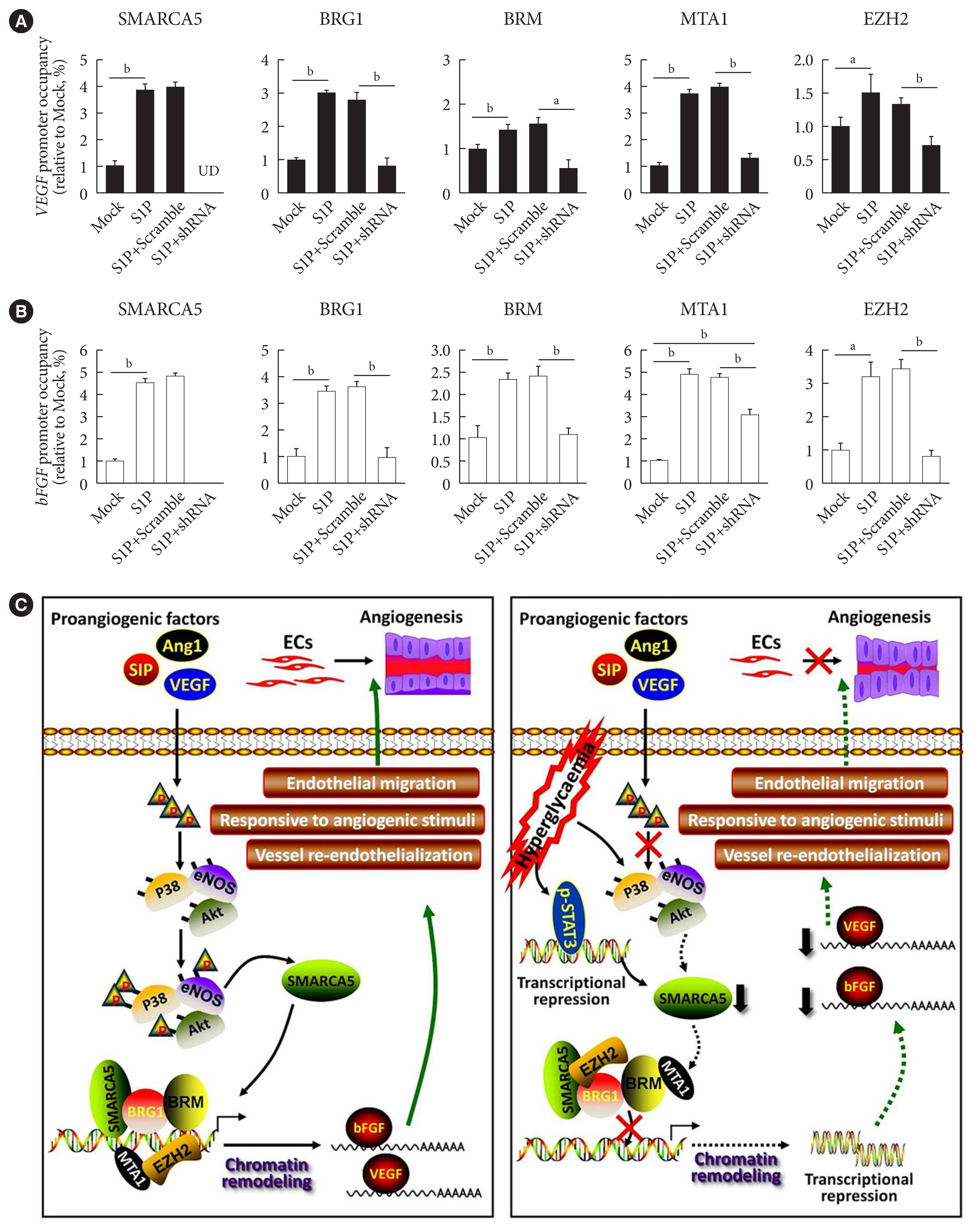
- SMARCA5 depletion disrupts transcriptional homeostasis in stimulated ECs
- To further dissect the mechanism by which SMARCA5 depletion affected the activation of the pro-angiogenic factors, we performed chromatin immunoprecipitation (ChIP)-qPCR assays, using antibodies against SMARCA5 and two core subunits of the SWI/SNF complex (namely Brahma-related gene 1 [BRG1] and Brahma homologue [BRM]), in HUVECsSMARCA5−/− and control HUVECs. Upon SMARCA5 depletion, the associations between BRG1/BRM and VEGF promoter regions were significantly decreased, even in the presence of S1P stimulation. Chromatin remodeling also accompanies histone acetylation and methylation [31]. Intriguingly, SMARCA5 depletion also resulted in the dissociation of both metastasis-associated protein 1 (MTA1; a well-known oncogenic chromatin modifier recruiting histone deacetylases) [32] and the the histone methyltransferase enhancer of zeste 2 polycomb repressive complex 2 subunit (EZH2) from the activated VEGF promoter regions in S1P-stimulated HUVECsSMARCA5−/− (Fig. 7A). Similar disassociation between these chromatin remodelers and promoter regions of activated bFGF gene was also observed in S1P-stimulated HUVECsSMARCA5−/− (Fig. 7B). Thus, SMARCA5 loss triggered the reassembly of the SWI/SNF complex as well as histone modifiers and, subsequently, chromatin remodeling to affect transcriptional events in ECs.
RESULTS
- Due to their unique anatomical locations in the cardiovascular system, ECs are constantly subjected to an array of mechanical and biochemical cues [33]. ECs sense, respond, and adapt to these altered cues by specialized mechanisms of mechanosensing and mechanotransduction, thus resulting in qualitative and quantitative differences in their gene expression [34]. To this end, the emerging field of epigenetics is offering a novel perspective to inspect how endothelial dysfunction is regulated through chromatin remodeling under different physiological and pathological conditions. In fact, compelling evidence has documented a sophisticated regulatory program, involving multiple subunits of the SWI/SNF chromatin-remodeling complex, in cardiovascular development and disease. For instance, a conditional mutation of Brg1 in developing ECs results in abnormal vascular remodeling of the extraembryonic yolk sac, thus leading to embryonic lethality [35]. Likewise, Baf180 constitutive mutants have defective coronary vessel development around the ventricle and within the myocardium, which is likely caused by a problem with the epithelial-to-mesenchymal transition and the migration of epicardial cells [36]. More importantly, in concert with Krüppel-like factor 4 (KLF4) transcription factor, the SWI/SNF complex has been shown to modulate accessibility at enhancer sites that promote expression of homeostatic endothelial genes in physiological laminar shear stress-stimulated ECs [37]. Thus, the SWI/SNF complex plays a crucial role in both early vascular development and adult tissue angiogenesis. Our current dataset extends these understandings by disclosing a novel role of SMARCA5 in ECs, as a key effector of the pro-angiogenic signaling-regulatory machinery, with a physiological function in the positive control of expression of pro-angiogenic factors (i.e., VEGF and bFGF) (Fig. 7C). Considering that hyperglycemia-induced decrease in VEGF expression renders ECs susceptible to damage in diabetes [38,39], repression of SMARCA5 by hyperglycemia-induced oxidative stress may mediate, at least in part, the vascular dysfunction caused by diabetes.
- Although the mechanisms whereby SMARCA5 expression is inhibited in ECs from diabetic mice remain to be defined, several regulators have been so far reported to determine the level of SMARCA5 expression. For example, in response to oxidative damage, SMARCA5 is recruited by 8-oxoguanine DNA glycosylase (OGG1) to interact with oxidative DNA damage sites and double-strand breaks, thus functioning as a potent oncogene in human colorectal cancer [40]. A similar activation of SMARCA5 signaling seems to take place also in glioblastoma, where SMARCA5 and the nucleosome remodeling (NuRD) complex are rapidly recruited to sites of DNA damage through SMARCA5 association with poly(ADP-ribose) polymerase 1 (PARP1) in tumor cells under constant oxidative stress [41]. Altogether, this evidence attests a sustained regulatory action of SMARCA5 in the control of genome integrity by regulating signaling and repair after oxidative damage. Interestingly, our molecular biological data demonstrated that oxidative stress may negatively regulate SMARCA5 transcription through a STAT3-dependent pathway in ECs (Fig. 5). Because ROS-mediated activation of STAT3 signaling aggravates endothelial dysfunction in diabetes [42], and because disturbance in redox homeostasis provokes genome-wide changes in transcriptional landscape and chromatin accessibility [43], we reason that SMARCA5 may function as an important converging point linking oxidative stress, transcriptional reprogramming, and endothelial dysfunction in diabetes. Moreover, miR-205 and miR-151-5p have been shown previously to inhibit SMARCA5 expression and negatively regulates its function in different cell types [44,45]. There is increasing evidence that miRNAs are responsible for fine-tuning the gene expression of important targets in the metabolic syndrome and T2DM [46], and deregulation of miRNA function (e.g., up-regulation of miR-205 or miR-151-5p) precedes and predicts endothelial dysfunction, thus contributing essentially to pathogenesis or progression of diabetes [46,47]. Hence, deregulation of oxidative stress or distinct miRNAs may at least partially explain the attenuated expression of SMARCA5 in diabetic ECs.
- While biochemical tests involving scratch migration, transwell migration and in vitro tube formation assays, already suggested a functional involvement of SMARCA5 in controlling pro-angiogenic response and subsequent angiogenesis, conclusive demonstration of the primary site and physiological relevance of such SMARCA5 regulatory cascade stemed from our studies using in vivo vasculogenesis assay, as well as Ad-SMARCA5 incorporated hydrogel-treated wound healing model. Ectopic overexpression of SMARCA5 significantly promoted vascularization in implants derived from ECFCs and hDFs (Fig. 3A–C). These data would suggest alternative modes of action of SMARCA5 in the tuning of function of EPCs, in line with recent mounting evidence demonstrating that SMARCA5-mediated epigenetic programming facilitates development of HSPCs [14]. It is increasingly recognized that specialized subsets of ECs carry out unique functions in specific organs of the vascular tree. Perhaps the most striking example of this specialization is the ability to contribute to the generation of a distinct population of “hemogenic” ECs in embryo, which ultimately transforms irreversibly into HSPCs [48]. Therefore, the available data collectively suggest that SMARCA5-mediated epigenetic modification may be functionally involved in control of the endothelial-to-hematopoietic transition. Additionally, diabetic mice treated by Ad-SMARCA5 incorporated hydrogel displayed enhanced wound healing in vivo, together with advanced reepithelialization and augmented subcutaneous cell proliferation (Fig. 4D–I). A similar acceleration of cell proliferation seems to occur also in HSPCs [49] and breast cancer [10]. Considering that the first step in angiogenesis takes place by the formation of a new sprout, off of the existing vasculature, mediated by tip cell migration and stalk cell proliferation [50], these findings together support a critical role of SMARCA5 in the regulation of multiple aspects of endothelial function (i.e., cell proliferation, migration, angiogenic response, and reendothelialization).
- VEGF is key mediator of angiogenesis in both embryonic development and cancer development, and is important in wound healing in adults [51]. However, the existence of multiple targets of VEGF in different contexts obscures the relationship among upstream regulators of VEGF. Our data set the grounds of this puzzle by identifying VEGF as the man downstream effector of SMARCA5 signaling during angiogenesis (Fig. 4). By using ChIP-qPCR assay, we further showed that SMARCA5 depletion disrupts transcriptional homeostasis in stimulated ECs (Fig. 5). More importantly, the activation of the pro-angiogenic transcriptome is probably due to both direct and indirect chromatin-remodeling mechanisms. On one hand, SMARCA5 depletion resulted in the dissociation of the SWI/SNF complex from DNA, resulting in the activation of pro-angiogenic genes following direct chromatin remodeling. On the other hand, SMARCA5 loss led to a change in histone modifications by the dissociation of MTA1 and EZH2 from chromatin, which triggered indirect chromatin remodeling to affect pro-angiogenic gene transcription. These findings thus support the diversity of functional mechanisms of SMARCA5 during angiogenesis.
- Of note, accumulated data have pointed out particular and unique biological characteristics of SMARCA5. In acute myeloid leukemia, SMARCA5 is oncogenic by virtue of stimulating cell proliferation and chromatid cohesion [8]. Similarly, a first-in-class SMARCA5/CHD4 inhibitor ED2-AD101 has been shown to bear therapeutic efficacy in regulating platinum sensitivity through MDR1 expression in ovarian cancer [52]. Thus, modulation of SMARCA5 in transformed and non-transformed cells (e.g., ECs) would thus have opposing effects. These striking observations stress the need for caution in therapeutic endeavors aimed at SMARCA5 in this and other contexts. Future studies using unbiased genome-wide examination of signaling pathways are warranted to provide more valuable information for a better understanding of the molecular mechanisms responsible for SMARCA5-potentiated changes in the transcriptional landscape following diabetic induction.
- In conclusion, the present study supports the notion that attenuation of SMARCA5 expression in ECs mediates endothelial dysfunction and may thereby exacerbate vascular complications in diabetes. Our findings therefore have translational potential, as they open up the possibility for the design of novel pharmacological strategies, targeting SMARCA5, for the management of metabolic disorders.
DISCUSSION
SUPPLEMENTARY MATERIALS
Supplementary Fig. 1.
Supplementary Table 1.
-
Acknowledgements
- We are grateful to participants of this study who made this work possible.
ACKNOWLEDGMENTS
-
CONFLICTS OF INTEREST
No potential conflict of interest relevant to this article was reported.
-
AUTHOR CONTRIBUTIONS
Conception or design: J.J.
Acquisition, analysis, or interpretation of data: J.W., H.Z., J.S., S.Z., J.J.
Drafting the work or revising: J.W., J.J.
Final approval of the manuscript: J.J.
-
FUNDING
None
NOTES

- 1. Knapp M, Tu X, Wu R. Vascular endothelial dysfunction, a major mediator in diabetic cardiomyopathy. Acta Pharmacol Sin 2019;40:1-8.ArticlePubMedPMCPDF
- 2. Yang C, Eleftheriadou M, Kelaini S, Morrison T, Gonzalez MV, Caines R, et al. Targeting QKI-7 in vivo restores endothelial cell function in diabetes. Nat Commun 2020;11:3812.ArticlePubMedPMCPDF
- 3. Aiello LP, Wong JS. Role of vascular endothelial growth factor in diabetic vascular complications. Kidney Int Suppl 2000;77:S113-9.ArticlePubMed
- 4. Kolluru GK, Bir SC, Kevil CG. Endothelial dysfunction and diabetes: effects on angiogenesis, vascular remodeling, and wound healing. Int J Vasc Med 2012;2012:918267.ArticlePubMedPMCPDF
- 5. Menon DU, Kirsanov O, Geyer CB, Magnuson T. Mammalian SWI/SNF chromatin remodeler is essential for reductional meiosis in males. Nat Commun 2021;12:6581.ArticlePubMedPMCPDF
- 6. Thakur S, Cahais V, Turkova T, Zikmund T, Renard C, Stopka T, et al. Chromatin remodeler smarca5 is required for cancer-related processes of primary cell fitness and immortalization. Cells 2022;11:808.ArticlePubMedPMC
- 7. Jevtic Z, Matafora V, Casagrande F, Santoro F, Minucci S, Garre M, et al. SMARCA5 interacts with NUP98-NSD1 oncofusion protein and sustains hematopoietic cells transformation. J Exp Clin Cancer Res 2022;41:34.ArticlePubMedPMCPDF
- 8. Zikmund T, Paszekova H, Kokavec J, Kerbs P, Thakur S, Turkova T, et al. Loss of ISWI ATPase SMARCA5 (SNF2H) in acute myeloid leukemia cells inhibits proliferation and chromatid cohesion. Int J Mol Sci 2020;21:2073.ArticlePubMedPMC
- 9. Cui T, Bell EH, McElroy J, Liu K, Sebastian E, Johnson B, et al. A novel miR-146a-POU3F2/SMARCA5 pathway regulates stemness and therapeutic response in glioblastoma. Mol Cancer Res 2021;19:48-60.ArticlePubMedPDF
- 10. Jin Q, Mao X, Li B, Guan S, Yao F, Jin F. Overexpression of SMARCA5 correlates with cell proliferation and migration in breast cancer. Tumour Biol 2015;36:1895-902.ArticlePubMedPDF
- 11. Ai C, Ma G, Deng Y, Zheng Q, Gen Y, Li W, et al. Nm23-H1 inhibits lung cancer bone-specific metastasis by upregulating miR-660-5p targeted SMARCA5. Thorac Cancer 2020;11:640-50.ArticlePubMedPMCPDF
- 12. Gigek CO, Lisboa LC, Leal MF, Silva PN, Lima EM, Khayat AS, et al. SMARCA5 methylation and expression in gastric cancer. Cancer Invest 2011;29:162-6.ArticlePubMed
- 13. Ding Y, Li Y, Zhao Z, Cliff Zhang Q, Liu F. The chromatin-remodeling enzyme Smarca5 regulates erythrocyte aggregation via Keap1-Nrf2 signaling. Elife 2021;10:e72557.ArticlePubMedPMCPDF
- 14. Ding Y, Wang W, Ma D, Liang G, Kang Z, Xue Y, et al. Smarca5-mediated epigenetic programming facilitates fetal HSPC development in vertebrates. Blood 2021;137:190-202.ArticlePubMedPMCPDF
- 15. Dong YS, Hou WG, Li Y, Liu DB, Hao GZ, Zhang HF, et al. Unexpected requirement for a binding partner of the syntaxin family in phagocytosis by murine testicular Sertoli cells. Cell Death Differ 2016;23:787-800.ArticlePubMedPMCPDF
- 16. Chai X, Yan J, Gao Y, Jin J. Endothelial HNF4α potentiates angiogenic dysfunction via enhancement of vascular endothelial growth factor resistance in T2DM. J Cell Biochem 2019;120:12989-3000.ArticlePubMedPDF
- 17. Hao JS, Zhu CJ, Yan BY, Yan CY, Ling R. Stimulation of KLF14/PLK1 pathway by thrombin signaling potentiates endothelial dysfunction in type 2 diabetes mellitus. Biomed Pharmacother 2018;99:859-66.ArticlePubMed
- 18. He K, Qu H, Wang H, Zhang S, Qian XH, Li W. Regulated and functional expression of the corepressor MTA3 in rodent testis. Endocrinology 2016;157:4400-10.ArticlePubMed
- 19. Fadini GP, Sartore S, Albiero M, Baesso I, Murphy E, Menegolo M, et al. Number and function of endothelial progenitor cells as a marker of severity for diabetic vasculopathy. Arterioscler Thromb Vasc Biol 2006;26:2140-6.ArticlePubMed
- 20. Sawada N, Jiang A, Takizawa F, Safdar A, Manika A, Tesmenitsky Y, et al. Endothelial PGC-1α mediates vascular dysfunction in diabetes. Cell Metab 2014;19:246-58.ArticlePubMedPMC
- 21. Cheng S, Cui Y, Fan L, Mu X, Hua Y. T2DM inhibition of endothelial miR-342-3p facilitates angiogenic dysfunction via repression of FGF11 signaling. Biochem Biophys Res Commun 2018;503:71-8.ArticlePubMed
- 22. Niccoli S, Boreham DR, Phenix CP, Lees SJ. Non-radioactive 2-deoxy-2-fluoro-D-glucose inhibits glucose uptake in xenograft tumours and sensitizes HeLa cells to doxorubicin in vitro. PLoS One 2017;12:e0187584.ArticlePubMedPMC
- 23. Chen YH, Lin SJ, Lin FY, Wu TC, Tsao CR, Huang PH, et al. High glucose impairs early and late endothelial progenitor cells by modifying nitric oxide-related but not oxidative stress-mediated mechanisms. Diabetes 2007;56:1559-68.ArticlePubMedPDF
- 24. Qi X, Okamoto Y, Murakawa T, Wang F, Oyama O, Ohkawa R, et al. Sustained delivery of sphingosine-1-phosphate using poly(lactic-co-glycolic acid)-based microparticles stimulates Akt/ERK-eNOS mediated angiogenesis and vascular maturation restoring blood flow in ischemic limbs of mice. Eur J Pharmacol 2010;634:121-31.ArticlePubMed
- 25. Tie L, Li XJ, Wang X, Channon KM, Chen AF. Endothelium-specific GTP cyclohydrolase I overexpression accelerates refractory wound healing by suppressing oxidative stress in diabetes. Am J Physiol Endocrinol Metab 2009;296:E1423-9.ArticlePubMedPMC
- 26. Marshall CD, Hu MS, Leavitt T, Barnes LA, Cheung AT, Malhotra S, et al. Sanativo wound healing product does not accelerate reepithelialization in a mouse cutaneous wound healing model. Plast Reconstr Surg 2017;139:343-52.ArticlePubMed
- 27. Fiorentino TV, Prioletta A, Zuo P, Folli F. Hyperglycemia-induced oxidative stress and its role in diabetes mellitus related cardiovascular diseases. Curr Pharm Des 2013;19:5695-703.ArticlePubMed
- 28. Ning P, Zhong JG, Jiang F, Zhang Y, Zhao J, Tian F, et al. Role of protein S in castration-resistant prostate cancer-like cells. Endocr Relat Cancer 2016;23:595-607.ArticlePubMed
- 29. Jin X, Zhang S, Ding T, Zhao P, Zhang C, Zhang Y, et al. Testicular Lmcd1 regulates phagocytosis by Sertoli cells through modulation of NFAT1/Txlna signaling pathway. Aging Cell 2020;19:e13217.ArticlePubMedPMCPDF
- 30. Wang GG, Wang YZ, Xie J, Huang CY, Kong ZL, Ding X, et al. Cyclic tensile forces enhance the angiogenic properties of HUVECs by promoting the activities of human periodontal ligament cells. J Periodontol 2021;92:159-69.ArticlePubMedPDF
- 31. Luo Q, Wu X, Chang W, Zhao P, Zhu X, Chen H, et al. ARID1A hypermethylation disrupts transcriptional homeostasis to promote squamous cell carcinoma progression. Cancer Res 2020;80:406-17.ArticlePubMedPDF
- 32. Liu J, Li C, Wang J, Xu D, Wang H, Wang T, et al. Chromatin modifier MTA1 regulates mitotic transition and tumorigenesis by orchestrating mitotic mRNA processing. Nat Commun 2020;11:4455.ArticlePubMedPMCPDF
- 33. Monteiro JP, Bennett M, Rodor J, Caudrillier A, Ulitsky I, Baker AH. Endothelial function and dysfunction in the cardiovascular system: the long non-coding road. Cardiovasc Res 2019;115:1692-704.ArticlePubMedPMCPDF
- 34. Kumar S. SWI/SNF (BAF) complexes: from framework to a functional role in endothelial mechanotransduction. Curr Top Membr 2021;87:171-98.ArticlePubMed
- 35. Griffin CT, Brennan J, Magnuson T. The chromatin-remodeling enzyme BRG1 plays an essential role in primitive erythropoiesis and vascular development. Development 2008;135:493-500.ArticlePubMedPMCPDF
- 36. Huang X, Gao X, Diaz-Trelles R, Ruiz-Lozano P, Wang Z. Coronary development is regulated by ATP-dependent SWI/SNF chromatin remodeling component BAF180. Dev Biol 2008;319:258-66.ArticlePubMed
- 37. Moonen JR, Chappell J, Shi M, Shinohara T, Li D, Mumbach MR, et al. KLF4 recruits SWI/SNF to increase chromatin accessibility and reprogram the endothelial enhancer landscape under laminar shear stress. Nat Commun 2022;13:4941.ArticlePubMedPMCPDF
- 38. Ishikura K, Misu H, Kumazaki M, Takayama H, Matsuzawa-Nagata N, Tajima N, et al. Selenoprotein P as a diabetes-associated hepatokine that impairs angiogenesis by inducing VEGF resistance in vascular endothelial cells. Diabetologia 2014;57:1968-76.ArticlePubMedPDF
- 39. Alhusban A, Alkhazaleh E, El-Elimat T. Silymarin ameliorates diabetes-induced proangiogenic response in brain endothelial cells through a GSK-3β inhibition-induced reduction of VEGF release. J Diabetes Res 2017;2017:2537216.ArticlePubMedPMCPDF
- 40. Xia L, Huang W, Bellani M, Seidman MM, Wu K, Fan D, et al. CHD4 has oncogenic functions in initiating and maintaining epigenetic suppression of multiple tumor suppressor genes. Cancer Cell 2017;31:653-68e7.ArticlePubMedPMC
- 41. McKenzie LD, LeClair JW, Miller KN, Strong AD, Chan HL, Oates EL, et al. CHD4 regulates the DNA damage response and RAD51 expression in glioblastoma. Sci Rep 2019;9:4444.ArticlePubMedPMCPDF
- 42. Yun JH, Park SW, Kim KJ, Bae JS, Lee EH, Paek SH, et al. Endothelial STAT3 activation increases vascular leakage through downregulating tight junction proteins: implications for diabetic retinopathy. J Cell Physiol 2017;232:1123-34.ArticlePubMedPDF
- 43. Levings DC, Lacher SE, Palacios-Moreno J, Slattery M. Transcriptional reprogramming by oxidative stress occurs within a predefined chromatin accessibility landscape. Free Radic Biol Med 2021;171:319-31.ArticlePubMedPMC
- 44. Tommasi S, Pinto R, Danza K, Pilato B, Palumbo O, Micale L, et al. miR-151-5p, targeting chromatin remodeler SMARCA5, as a marker for the BRCAness phenotype. Oncotarget 2016;7:80363-72.ArticlePubMedPMC
- 45. Wu Z, Tang H, Xiong Q, Liu D, Xia T, Liang H, et al. Prognostic role of microRNA-205 in human gynecological cancer: a meta-analysis of fourteen studies. DNA Cell Biol 2020;39:875-89.ArticlePubMed
- 46. Ouni M, Gottmann P, Westholm E, Schwerbel K, Jahnert M, Stadion M, et al. MiR-205 is up-regulated in islets of diabetes-susceptible mice and targets the diabetes gene Tcf7l2. Acta Physiol (Oxf) 2021;232:e13693.ArticlePubMedPDF
- 47. Zhang Q, Xiao X, Li M, Li W, Yu M, Zhang H, et al. Acarbose reduces blood glucose by activating miR-10a-5p and miR-664 in diabetic rats. PLoS One 2013;8:e79697.ArticlePubMedPMC
- 48. Sugden WW, North TE. Making blood from the vessel: extrinsic and environmental cues guiding the endothelial-to-hematopoietic transition. Life (Basel) 2021;11:1027.ArticlePubMedPMC
- 49. Kokavec J, Zikmund T, Savvulidi F, Kulvait V, Edelmann W, Skoultchi AI, et al. The ISWI ATPase Smarca5 (Snf2h) is required for proliferation and differentiation of hematopoietic stem and progenitor cells. Stem Cells 2017;35:1614-23.ArticlePubMedPMCPDF
- 50. Norton KA, Popel AS. Effects of endothelial cell proliferation and migration rates in a computational model of sprouting angiogenesis. Sci Rep 2016;6:36992.ArticlePubMedPMCPDF
- 51. Carmeliet P. VEGF as a key mediator of angiogenesis in cancer. Oncology 2005;69(Suppl 3):4-10.ArticlePubMedPDF
- 52. Oyama Y, Shigeta S, Tokunaga H, Tsuji K, Ishibashi M, Shibuya Y, et al. CHD4 regulates platinum sensitivity through MDR1 expression in ovarian cancer: a potential role of CHD4 inhibition as a combination therapy with platinum agents. PLoS One 2021;16:e0251079.ArticlePubMedPMC
 PubReader
PubReader ePub Link
ePub Link Cite
Cite