- Current
- Browse
- Collections
-
For contributors
- For Authors
- Instructions to authors
- Article processing charge
- e-submission
- For Reviewers
- Instructions for reviewers
- How to become a reviewer
- Best reviewers
- For Readers
- Readership
- Subscription
- Permission guidelines
- About
- Editorial policy
Articles
- Page Path
- HOME > Diabetes Metab J > Volume 45(6); 2021 > Article
-
ReviewBasic Research Mitochondrial TFAM as a Signaling Regulator between Cellular Organelles: A Perspective on Metabolic Diseases
-
Jin-Ho Koh1
 , Yong-Woon Kim1, Dae-Yun Seo2, Tae-Seo Sohn3
, Yong-Woon Kim1, Dae-Yun Seo2, Tae-Seo Sohn3 -
Diabetes & Metabolism Journal 2021;45(6):853-865.
DOI: https://doi.org/10.4093/dmj.2021.0138
Published online: November 22, 2021
1Department of Physiology, Yeungnam University College of Medicine, Daegu, Korea
2Cardiovascular and Metabolic Disease Center, Smart Marine Therapeutic Center, Department of Physiology, College of Medicine, Inje University, Busan, Korea
3Department of Internal Medicine, Uijeongbu St. Mary’s Hospital, College of Medicine, The Catholic University of Korea, Seoul, Korea
-
Corresponding authors: Jin-Ho Koh Department of Physiology, Yeungnam University College of Medicine, 170 Hyeonchungro, Nam-gu, Daegu 42415, Korea E-mail: jinhokoh@yu.ac.kr
-
Tae-Seo Shon Department of Internal Medicine, Uijeongbu St. Mary’s Hospital, College of Medicine, The Catholic University of Korea, 271 Cheonbo-ro, Uijeongbu 11765, Korea E-mail: imsts@catholic.ac.kr
Copyright © 2021 Korean Diabetes Association
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- Tissues actively involved in energy metabolism are more likely to face metabolic challenges from bioenergetic substrates and are susceptible to mitochondrial dysfunction, leading to metabolic diseases. The mitochondria receive signals regarding the metabolic states in cells and transmit them to the nucleus or endoplasmic reticulum (ER) using calcium (Ca2+) for appropriate responses. Overflux of Ca2+ in the mitochondria or dysregulation of the signaling to the nucleus and ER could increase the incidence of metabolic diseases including insulin resistance and type 2 diabetes mellitus. Mitochondrial transcription factor A (Tfam) may regulate Ca2+ flux via changing the mitochondrial membrane potential and signals to other organelles such as the nucleus and ER. Since Tfam is involved in metabolic function in the mitochondria, here, we discuss the contribution of Tfam in coordinating mitochondria-ER activities for Ca2+ flux and describe the mechanisms by which Tfam affects mitochondrial Ca2+ flux in response to metabolic challenges.
- Tissues that are very active in energy metabolism are more likely to face metabolic challenges from the bioenergetic substrate and are susceptible to mitochondrial diseases [1]. Several genes from both the nucleus and mitochondria are required for mitochondrial formation and maturity. Thus, intercommunication between the mitochondria and nucleus is critical for highly metabolic tissues such as skeletal muscle, heart, liver, and adipose tissue.
- The mitochondria are heavily involved in diabetes, insulin resistance, complications derived from diabetes [2], and cellular calcium (Ca2+) flux, an important process for mitochondrial function and metabolic diseases since Ca2+ signaling regulates various events related to cell death and energy metabolism [3-7]. Mitochondrial metabolic dysfunctions induced by insulin resistance are linked to abnormal Ca2+ flux and high reactive oxygen species (ROS) levels. The metabolic functions that regulate Ca2+ flux are also associated with glucose uptake and metabolism [4]. The nucleus responds to Ca2+ signals, yet the endoplasmic reticulum (ER) dynamically interacts with the mitochondria and regulates Ca2+ signaling. This process is essential for several metabolic processes and physiological functions [8], and is important for regulating normal mitochondrial biogenesis [5,9] and glucose metabolism [4].
- The nucleus encodes the mitochondrial transcription factor A (TFAM), which activates transcription in the mitochondria and transmits a signal to the nucleus based on the mitochondrial state by regulating Ca2+ levels. It has been shown that it also prevents high-fat diet-induced insulin resistance [10]. Tfam-mutant mice developed diabetes and exhibited mitochondrial deoxyribonucleic acid (mtDNA) depletion, deficient oxidative phosphorylation, and abnormal mitochondrial structure [9]. This suggests that TFAM is involved in cellular metabolic function, possibly involving Ca2+ signaling.
- This review focuses on how mitochondrial TFAM regulates Ca2+ signaling and how dysregulation of Ca2+ by the mitochondria leads to metabolic disease in type 2 diabetes mellitus (T2DM).
INTRODUCTION
- Mitochondria play a key role in cellular physiology, and are responsible for producing cellular energy and essential metabolites, along with regulating apoptosis [11]. These functions are dependent on gene expression in both the mitochondria and nucleus and are regulated by communication between mitochondria and other organelles.
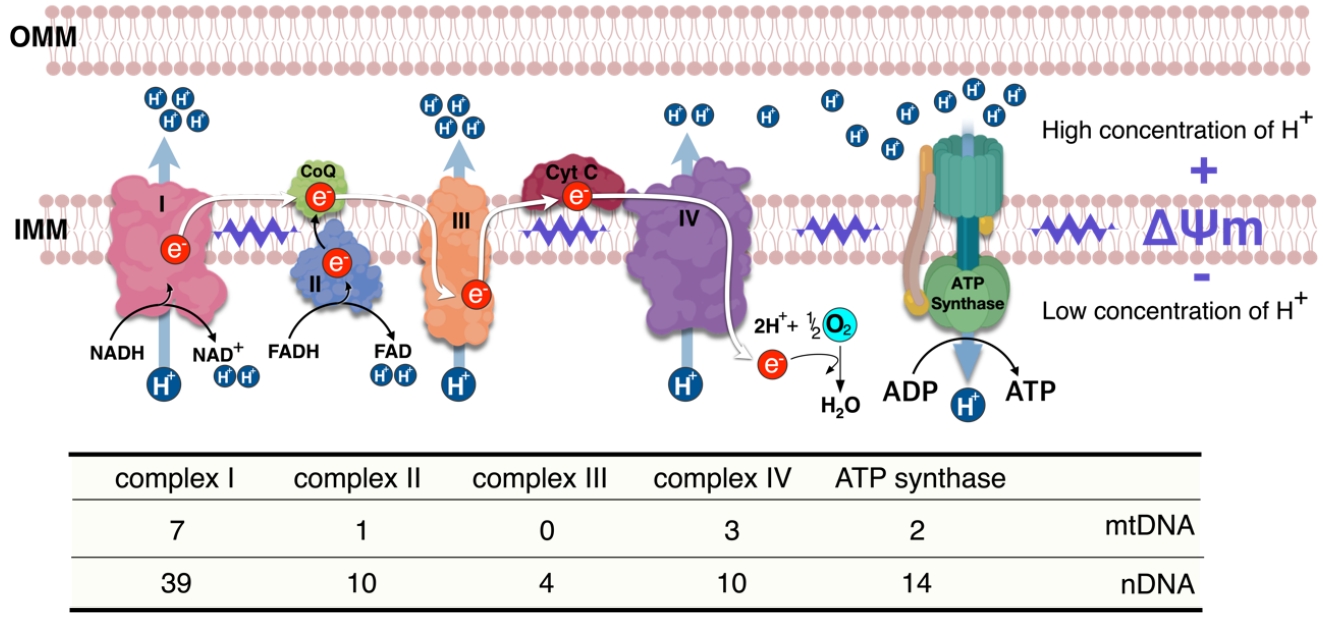
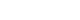
- Biosynthesis of each electron transport chain (ETC) complex in the mitochondria is jointly regulated by the nucleus and mtDNA. Since several genes are required for mitochondrial biogenesis (Fig. 1), TFAM is an important regulator between the mitochondria and nucleus, since Tfam is expressed from the nucleus but acts on the mitochondria. TFAM regulates transcription of the 13 genes for ETC protein, 22 for transfer RNAs, and two for ribosomal RNAs encoded by mtDNA [12,13]. TFAM binds to mtDNA and enhances transcription in association with mitochondrial RNA polymerase and either mitochondrial transcription factor B1 (TFB1M) or B2 (TFB2M) [14]. In addition to binding to the specific region of the promoter, TFAM binds nonspecifically to random sites on mtDNA [15,16] which enhances the stabilization and maintenance of the mitochondrial chromosome [15-17] as well as regulation of mtDNA copy number [18]. Interestingly, since TFAM overexpression has been shown to increase mtDNA without alteration of respiratory capacity and mitochondrial biogenesis [10], it seems that the mechanisms underlying mitochondrial biogenesis and maintenance of mtDNA by TFAM are separately regulated. Moreover, TFAM overexpression increases the nuclear expression of various factors associated with Ca2+ signaling and glucose metabolism in mouse skeletal muscle [10]. TFAM is also involved in mitochondrial function including lower ROS via enhancing antioxidants, 5’ adenosine monophosphate-activated protein kinase (AMPK) activation, and mitochondrial uncoupling as well as regulation of membrane potential [10]. Therefore, intercommunication between the mitochondria and nucleus is closely regulated to cope with a sudden change in cellular energy challenges, with TFAM possibly playing an important role in this interaction.
MITOCHONDRIA AND TFAM
- Signaling from the nucleus can regulate mitochondrial gene expression or mtDNA replication in response to cellular metabolic challenges or environmental signals [19], via TFAM. Nuclear respiratory factor 1 (NRF-1) can bind specific promoters of various nuclear genes required for mitochondrial respiratory function [20], which coordinates the expression of respiratory subunits with the mitochondrial transcriptional system [13]. NRF-1 also binds and activates the Tfam promoters [21], thereby inducing a cellular signaling cascade in skeletal muscle in response to exercise training [22,23]. In this context, Ca2+ treatment in muscle cell increases TFAM and NRF-1 protein levels as well as mitochondrial biogenesis [5,9], suggesting Ca2+ as a signaling molecule between cellular organelles. Redox reactions are activated in the mitochondria during exercise-induced oxidative phosphorylation and the incidence of NRF-1 binding to the TFAM promoter is increased under pro-oxidant conditions. However, Tfam expression is inhibited by deactivated NRF-1 [24], suggesting that Tfam expression mediated by NRF-1 is regulated under activated redox conditions. Interestingly, mtDNA depletion induces an increase in both along with oxidative stress [25]. NRF-1 and Tfam mRNAs are also increased in response to mitochondrial lipopolysaccharide-induced oxidative damage [26], suggesting that TFAM is increased to mitigate these cellular conditions, which, along with NRF-1, have been shown to decrease ROS via enhanced antioxidant and mitochondrial uncoupling [10].
- Taken together, when the cellular environment undergoes metabolic challenge, including exercise or metabolic alteration, NRF-1 and TFAM are upregulated in the nucleus followed by TFAM translocation into the mitochondria to mitigate this change by regulating mitochondrial oxidative phosphorylation and redox valence.
- Intercommunication between mitochondria and ER for intracellular Ca2+ flux
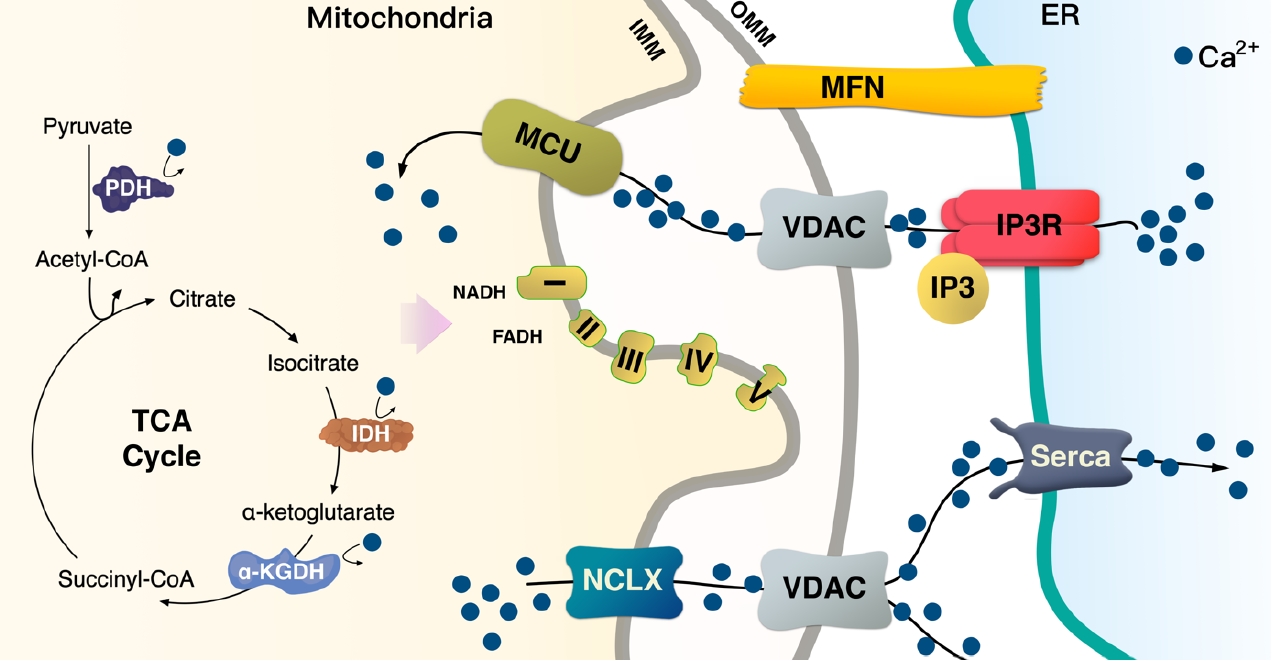
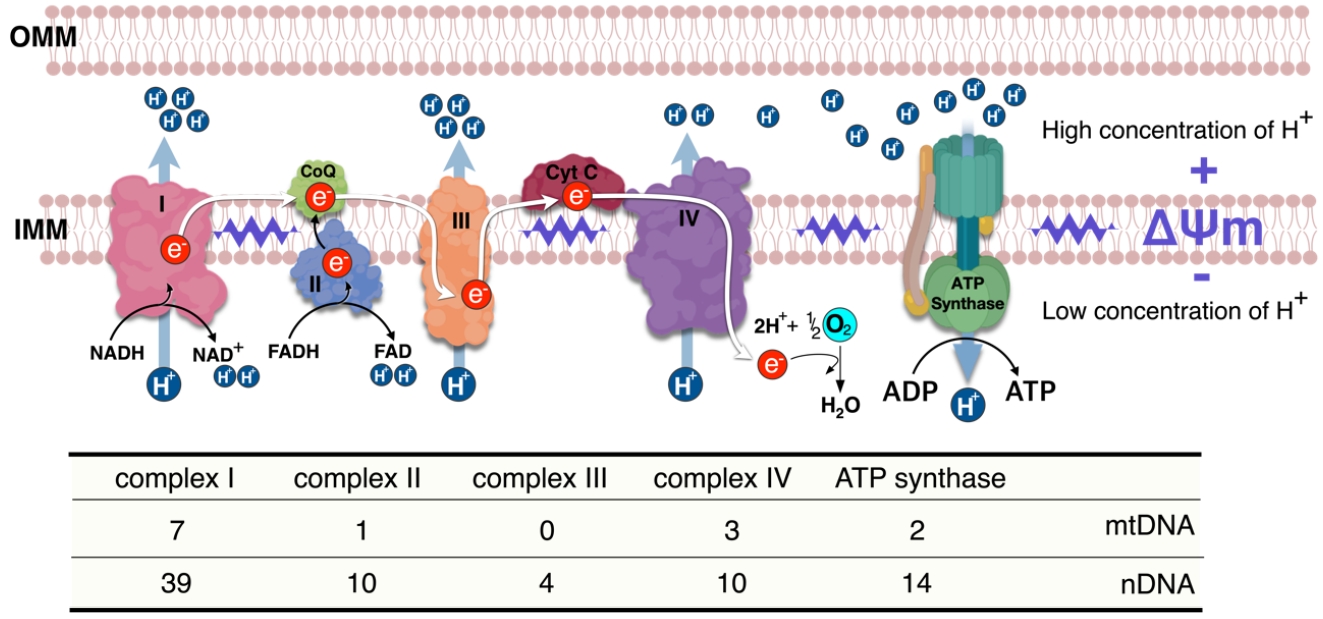
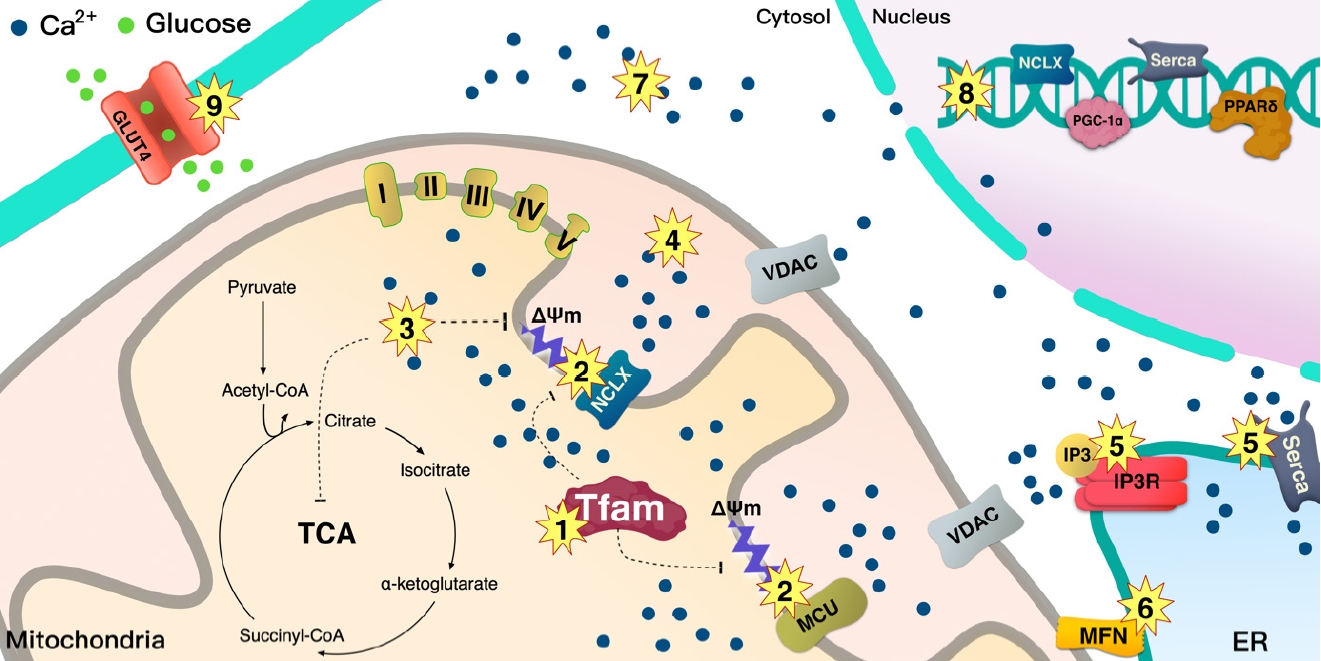
- Ca2+ signaling and flux regulate numerous cellular physiological processes, including neuronal excitability, muscle contraction, nuclear gene expression, and mitochondrial integrity, function, and dynamics [27]. Accumulated data have shown that mitochondrial Ca2+ content is low under basal conditions, while increases in cytosolic free Ca2+ in response to various agents (nutrients, hormones, neurotransmitters, etc.) elevates mitochondrial Ca2+ levels [28,29]. This enhances the activity of tricarboxylic acid (TCA) cycle dehydrogenase (pyruvate-, isocitrate-, and α-ketoglutarate dehydrogenase) required for oxidative phosphorylation [30]. Thus, activation of oxidative metabolism via enhanced Ca2+ flux can increase the supply of redox cofactors, such as a reduced form of nicotinamide adenine dinucleotide (NAD) and flavin adenine dinucleotide (FAD) to drive ETC and adenosine triphosphate (ATP) synthesis (Fig. 2).
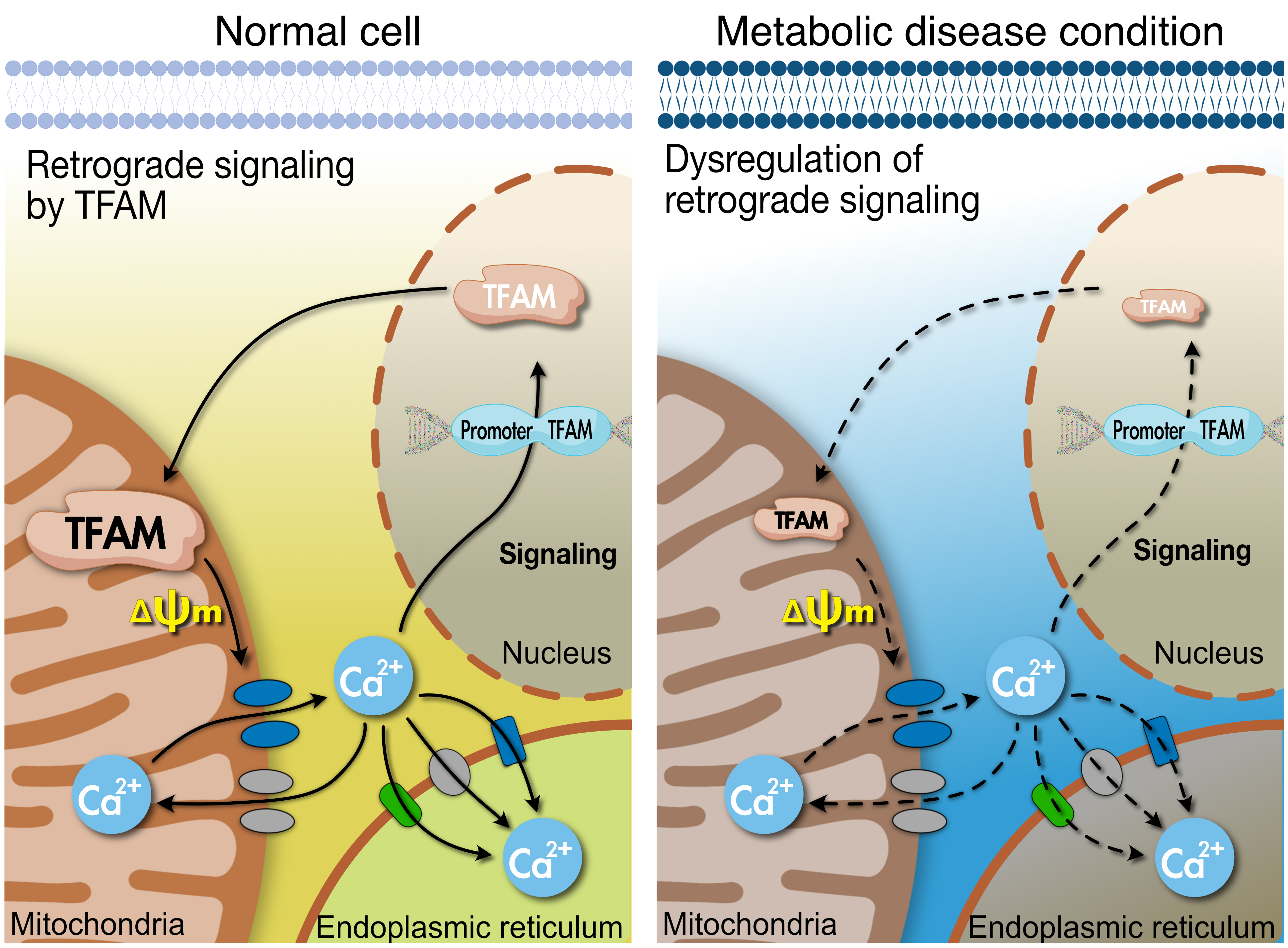
- TFAM regulates the mitochondrial membrane potential (ΔΨm) and increases calcium/calmodulin-dependent protein kinase kinase β (CaMKKβ) [10]. In turn, ΔΨm can regulate cellular Ca2+ flux [31], while CaMKKβ is activated in a calcium-dependent manner [32]. Thus, TFAM-driven ΔΨm controls Ca2+ flux in the cell via various Ca2+ channels. Perturbation of TFAM expression cannot protect mtDNA [18], while damaged mtDNA has been reported to increase ER stress [33]. This suggests that TFAM may be involved in the mitochondria-ER interactions.
- The lumen of the ER is the main storage site of Ca2+ and is constantly refilled via the sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) pump [34,35]. When skeletal muscle contraction is initiated, cytosolic Ca2+ concentration is rapidly increased by 3 to 4-fold, with the sarcoplasmic reticulum (SR), an ER in skeletal muscle, optimizing the coupling of excitation and contraction of muscle fibers. SR has channels for efflux of Ca2+ into the cytosol in response to depolarization, while SERCA transports cytosolic Ca2+ in an ATP-dependent manner into the ER lumen, thereby terminating the contraction. The close link between the mitochondria and SR/ER allows for rapid and potent local Ca2+ signaling (Fig. 2) [6,36].
- Mitochondrial and endoplasmic reticulum contacts (MERC) orchestrate cellular physiological functions such as mitochondrial Ca2+ signaling and dynamics. Under physiological stimulation requiring intercommunication between mitochondria and ER, mitofusin 2 (MFN2) tethers to stabilize both organelles [31,37,38]. Meanwhile, inositol 1,4,5-trisphosphate (IP3) binds to the IP3 receptor (IP3R), triggering Ca2+ release from the ER through IP3R, which can directly release Ca2+ into the cytosol or mitochondria [36]. Released Ca2+ from the ER enters the mitochondrial intermembrane space by first passing through the outer mitochondrial membrane via the voltage-dependent anion channel (VDAC) [39], then the mitochondrial calcium uniporter (MCU) transporter across the inner mitochondrial membrane [40,41]. TFAM is linked to Ca2+ regulation via MFN2 [42]. Ca2+ flux is essential for mitochondrial bioenergetic processes, which is linked to the ΔΨm (Figs. 1 and 2).
- TFAM regulates the mitochondrial and ER Ca2+ signaling to nucleus via ΔΨm
- Mitochondria are key organelles in Ca2+ flux regulation in cells. While Ca2+ flux in mitochondria is one of the most significant processes in regulating energy production and communication with other organelles, it mediates various pathologies associated with metabolic disease.
- Mitochondrial Ca2+ uptake is directly linked to mitochondrial bioenergetics, where depletion of mitochondrial ΔΨm abrogates mitochondrial Ca2+ uptake, and defects in the respiratory chain have been linked to the decreased ability of mitochondria to pump Ca2+. Energetic substrates derived by the TCA cycle are serially reduced to equivalents by ETC, and these redox reactions are coupled with protons pumping from the matrix into the intermembrane space [43]. The proton electrochemical gradient induced by ΔΨm and pH gradient is necessary to produce ATP (Fig. 1). Thus, ΔΨm can maintain mitochondrial Ca2+ uptake and physiological functions (Figs. 1 and 2) [44,45].
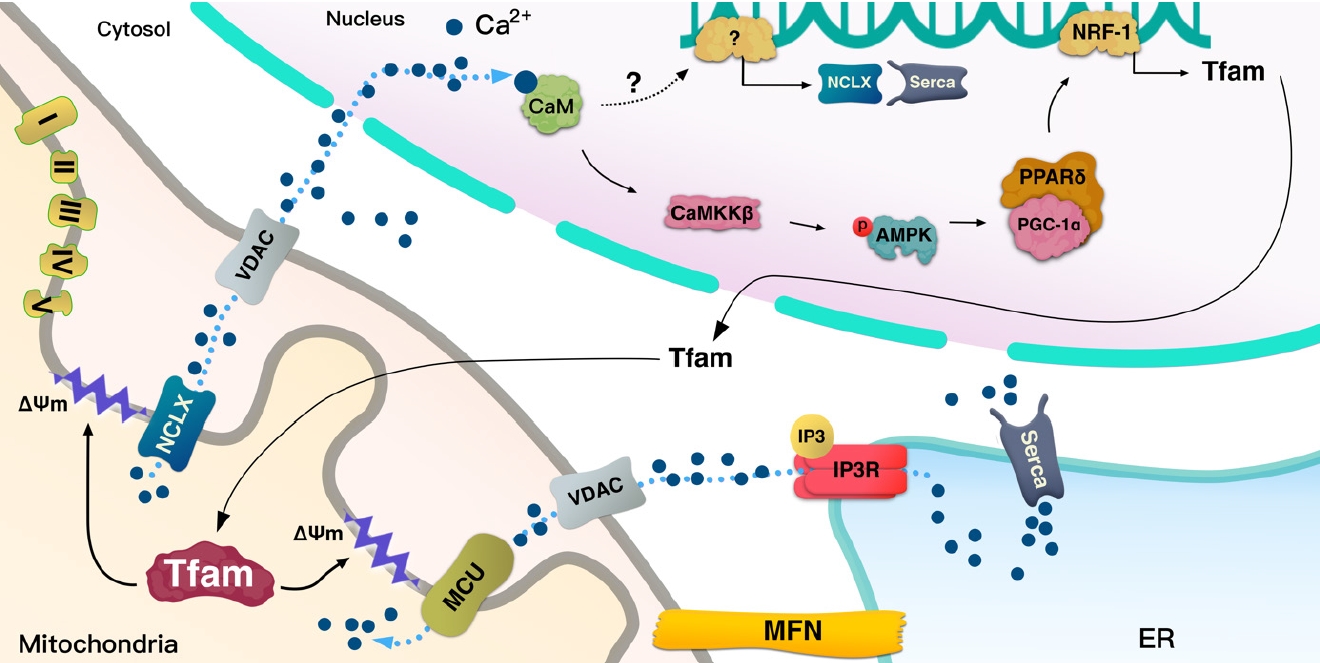
- Lack of TFAM decreases Serca2a expression from the nucleus [46], indicating that TFAM in the mitochondria signals to the nucleus to transcribe Serca2a and coordinate with ER to maintain Ca2+ homeostasis. The ER can regulate the Ca2+ efflux channels [36], which could affect cytosolic Ca2+ and force during contraction; hence, a reduced ER Ca2+ level in TFAM knockout muscle results in a decrease in cytosolic Ca2+ [47], which leads to a lower force in single fibers during contraction [47]. In TFAM knockout mice, an increase in mitochondrial Ca2+ occurs during repeated contraction [47], which may induce Ca2+ overload in the mitochondria and oxidative stress. In contrast with TFAM knockouts, Serca2 gene can enhance contractile function and restore electrical stability in a heart failure model caused by ER(SR) Ca2+ leak [48]. Watanabe et al. [46] have shown that TFAM regulates Serca2 gene transcription; however, it is not clear how TFAM in mitochondria regulates nuclear Serca2a transcription (Fig. 3).
- As mentioned above, mice with muscle-specific deletion of TFAM exhibit Ca2+ overload in the mitochondria, which might be caused by the low Ca2+ efflux into the cytosol by the mitochondria. MCU is the main channel transporting Ca2+ from the cytosol, and Na+/Ca2+ exchanger (NCLX) regulates efflux of Ca2+ from mitochondria, both of which are located at the inner mitochondrial membrane where they coordinate to regulate mitochondrial Ca2+ homeostasis. Mice with cardiac-specific deletion of TFAM showed how a failure in the coordination between MCU and NCLX in mitochondria can lead to cardiomyopathy [49]. TFAM deletion in cardiac muscle decrease Ca2+ efflux from mitochondria due to low NCLX gene and protein levels, with the reduced efflux potentially inducing mitochondrial Ca2+ overload [49]. Ca2+ flux through the MCU can be regulated by the voltage gradient across the inner mitochondrial membrane, while the opening probability of MCU is decreased based on ΔΨm depolarization [50]. Moreover, protonophore-induced depolarization of ΔΨm almost fully suppresses Ca2+ influx into the mitochondria [51]. These findings indicate that the large electrical driving force that arises from the negative potential across the inner ΔΨm is a major factor in regulating the influx of Ca2+ through MCU (Fig. 3).
- Depolarization of ΔΨm or lack of mtDNA prevents mitochondrial Ca2+ uptake, elevating the cytoplasmic Ca2+ level and leading to mitochondrial retrograde signaling into the nucleus to activate Ca2+-mediated transcription mechanisms involved in calcineurin and Ca2+/calmodulin-dependent protein kinase (CaMK) [44,45]. Human TFAM (hTFAM) transgenic mice exhibit mild ΔΨm uncoupling when fatty acids are used as a substrate for skeletal muscle mitochondria, despite mtDNA in skeletal muscle being increased by hTfam [10]. These alterations by hTfam result in an increase in mild uncoupling in ΔΨm, leading to an increase in CaMKKβ expression from the nucleus [10]. Therefore, TFAM regulates ΔΨm and mediates retrograde signaling to the nucleus by Ca2+ transmission. Interestingly, mild uncoupling of ΔΨm by hTFAM decreased ROS [10]. Since a lack of TFAM can mediate Ca2+ overload in the cytoplasm, it not only induces retrograde signaling to the nucleus but also increases ROS and apoptosis [45]. ROS produced by lack of TFAM in cells may be induced by an overload of Ca2+ [45]; however, hTFAM overexpression can decrease ROS and oxidative stress in tissue with mild uncoupling of ΔΨm [10]. Therefore, TFAM may mediate mild uncoupling of ΔΨm that regulates Ca2+ retrograde signaling to tightly control cellular ROS (Fig. 3).
- The nuclear genome encodes approximately 1,500 proteins that are necessary for mitochondrial function and integrity [52,53]. Intercommunication between mtDNA and the nuclear genome is necessary for mitochondrial biogenesis and normal function. To facilitate the interplay between the organelles, TFAM functions signaling transmitter to the mitochondria from the nucleus, and sends signals back to the nucleus according to the state of the mitochondria by regulating Ca2+. This signaling pathway is important for the regulation of normal mitochondrial biogenesis via peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), which orchestrates various transcription factors including NRF-1 and peroxisome proliferator-activated receptor delta (PPARδ) (Fig. 3).
- Intracellular Ca2+ binds CaM, thereby mediating various cellular functions [3,54]. In skeletal muscle, this Ca2+/CaM complex contract the muscle fiber [55]. The activation of Ca2+/CaMKKβ enhances AMPK activation [32], which facilitates mitochondrial biogenesis via enhanced expression of PGC-1α [56]. Muscle-specific hTFAM transgenic mice exhibit higher levels of CaMKKβ and activated AMPK [10]. To maintain and manage mtDNA as well as to regulate mitochondrial function, it is also necessary to increase the amount of TFAM as the mitochondrial number increases. It has been shown that TFAM overexpression in skeletal muscle enhances the expression of NRF-1, which is an upstream transcription factor for itself [10] and PPARδ expression. PPARδ regulates glucose transporter type 4 (GLUT4) expression and glucose uptake in muscle tissue [57]. A higher level of PGC-1α and PPARδ in tissue is beneficial in improving metabolic diseases such as insulin resistance and T2DM (Figs. 3 and 4).
- Ca2+ flux in metabolic disease
- Skeletal muscle, liver, and adipose are metabolic and insulin-sensitive tissues. Mitochondrial dysfunction in those tissues is associated with lower levels of TFAM in various metabolic diseases such as obesity, insulin resistance, and T2DM, which mediate abnormal Ca2+ flux.
- Role of MERC in diabetes
- Tethering between mitochondria and ER plays a key role in Ca2+ homeostasis, which regulates energy metabolism, transportation of lipids, and apoptosis [58]. The influx of Ca2+ mediates insulin-stimulated glucose uptake in skeletal muscle [4]. The authors reported that a decrease in Ca2+ influx by IP3R inhibition improved insulin-stimulated glucose uptake in skeletal muscle without AKT signaling [4]. In adipocytes, cytosolic Ca2+ levels were increased by insulin stimulation [59]. The inhibition of Ca2+ signaling in adipocytes [59] and lack of IP3R in primary rat cardiomyocytes [60] decreased GLUT4 translocation and glucose uptake upon insulin stimulation, respectively. Moreover, mitochondrial dysfunction [61], dysregulation of lipid and Ca2+ homeostasis [62], and ER stress [63] have been reported to be closely linked to insulin resistance in the liver. Palmitate is known to reduce insulin sensitivity; it has been shown that disruption of the interaction between mitochondria and ER by palmitate induces insulin resistance in human’s and mice’s hepatocytes. However, an increase in these associations between organelles can prevent palmitate-induced insulin resistance [64]. Loss of IP3R1 decreases MERC formation and induces mitochondrial dysfunction and insulin resistance; however, restoration of MERC has been shown to improve palmitate-induced insulin resistance in hepatocytes [64]. It has been reported that AKT phosphorylates the IP3R channel, resulting in decreased Ca2+ release capacity through IP3R [65,66]. Moreover, loss of IP3R activity has been shown to change the integrity of MERC [64]. Although defects in MERC have been suggested to play a role in insulin resistance and T2DM [37,38,42], MERC-mediated Ca2+ flux for glucose homeostasis is complicated and tightly regulated; hence, other specific mechanisms might be involved in this process. Thus, further studies are required to demonstrate the relationship between insulin resistance and MERC, as well as the mechanisms involved in this process. ER-associated mitochondrial division sites are spatially linked to mitochondrial nucleoids, which suggests a specific role for mitochondria-ER contacts in mtDNA maintenance [67]. ER-mitochondria contacts coordinate mtDNA synthesis with division to distribute newly replicated nucleoids into daughter mitochondria [68]. MFNs, proteins acting between mitochondria and ER [69], are also involved in metabolic diseases. Mitochondrial dysfunction is reportedly caused by depletion and point mutations of mtDNA in mice with muscle-specific deletion of MFN1 and MFN2 [69], while MFN2 expression in skeletal muscle is lower in patients with obesity or T2DM [37]. In contrast, overexpression of MFN2 has been shown to improve diet-induced insulin resistance [70], and MFN2 is necessary for normal glucose homeostasis [38]. Whether TFAM is directly involved in the process of forming or maintaining ER-mitochondria contacts remains to be determined, however, the loss of MFN1 and MFN2 decreases mtDNA that is maintained by TFAM [69], while PGC-1α, a transcription factor for Tfam, can increase the MFN2 expression levels via estrogen-related receptor-α (ERRα) [71]. Moreover, TFAM is involved in packaging mtDNA into a nucleoid [72-74]; hence, it is possible that TFAM indirectly participates in this process of MERC-mediated glucose homeostasis. hTFAM overexpression in skeletal muscle has been reported to prevent high-fat diet-induced insulin resistance along with preserving higher levels of mtDNA [10].
- SERCA tightly regulates cytosolic Ca2+, leading to glucose oxidation [75]. An increase in SERCA has been reported to improve diabetic cardiomyopathy [7,76]. A high-fat diet decreases Serca2a expression due to a lack of TFAM; however, overexpression of TFAM inhibits hydrogen peroxide-induced decrease of Serca2a mRNA [46]. An increase in SERCA2a by TFAM can reduce levels of cytoplasmic Ca2+ and calpain and mitochondrial apoptosis factor, thereby improving glucose uptake in cardiac muscle [77]. Increased expression of Serca improves metabolic syndrome [78].
- Role of mitochondrial Ca2+ regulation in T2DM
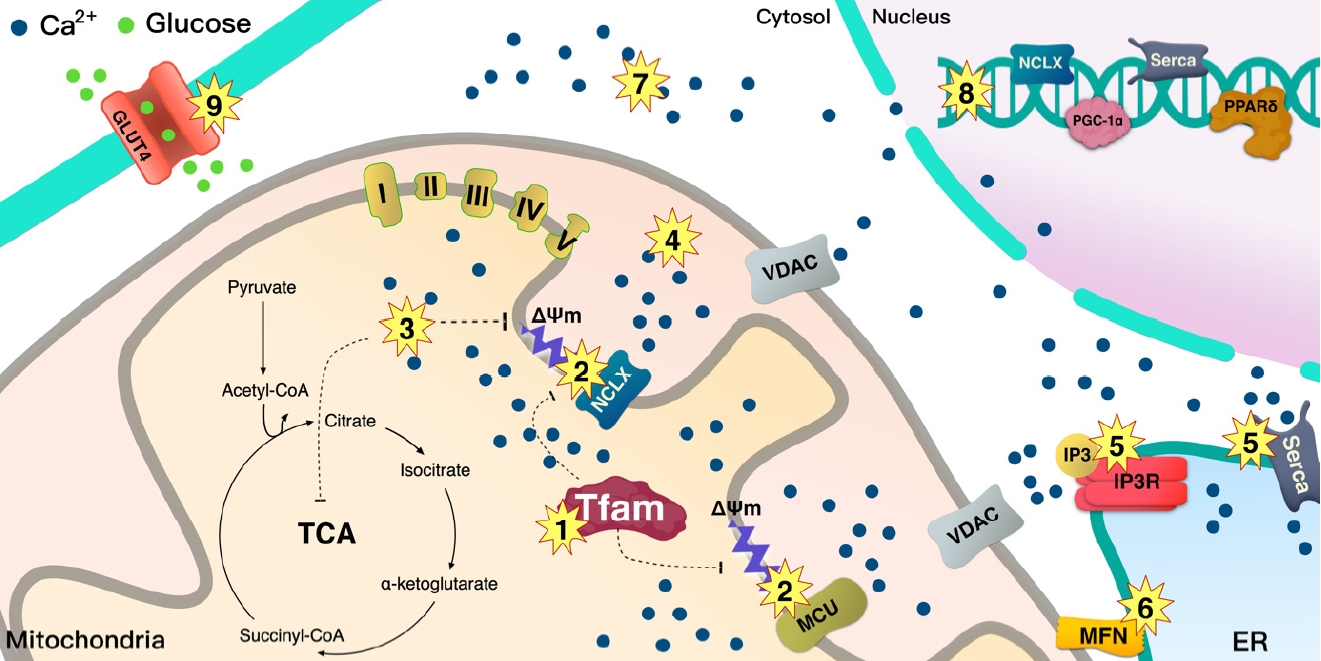
- The MCU complex consists of a pore-forming MCU subunit, a regulatory subunit, and mitochondrial Ca2+ uptake proteins 1-3 (MICU1-3), which regulate the channel activity [79-82]. Indeed, a splicing variant of muscle-specific MICU1 with higher Ca2+ affinity facilitates mitochondrial Ca2+ uptake and ATP production, which is required for muscle contraction [83], whereas a deletion of MCU in skeletal muscle in mice decreases muscle force, indicating that through regulating Ca2+ uptake, MCU plays a vital role in energy production for contraction [84]. Deletion of the muscle-specific MCU induced lower activity of pyruvate dehydrogenase (PDH), which is sufficient to shift the preference of the substrate toward fatty acids from carbohydrates [85] since PDH converts pyruvate to acetyl-CoA. In addition, loss of MCU causes defects in Ca2+-sensitive TCA cycle enzymes such as isocitrate and α-ketoglutarate dehydrogenases. Therefore, MCU-deleted mitochondria in skeletal muscle cannot sustain respiration without TCA cycle support, although MCU-deleted muscle mostly relies on fatty acids as substrate for mitochondrial respiration [85]. Thus, fatty acids do not completely oxidize and accumulate in muscle, leading to insulin resistance in skeletal muscle [86]. MCU is on the mitochondrial inner membrane that is dependent on ΔΨm, and hTFAM overexpression in skeletal muscle induces mild uncoupling of ΔΨm, which may change mitochondrial Ca2+ flux. This alteration shifts the preferred substrate for mitochondrial respiration from glucose to fatty acids [10]. However, the hTFAM transgenic model is different from the MCU deletion since muscle-specific hTFAM overexpression increases glucose uptake and prevents high-fat diet-induced insulin resistance (Figs. 3 and 4) [10].
- The NCLX plays a vital role in mitochondrial efflux into the cytosol [87]. A decrease in NCLX function leads to the accumulation of Ca2+ in mitochondria, while impaired NCLX function has been reported in diabetic rat hearts [87,88]. A previous study has shown that deletion of TFAM in mouse cardiomyocytes decreases NCLX transcription [49], and TFAM regulates ΔΨm in skeletal muscle; therefore, TFAM seems to clearly link to mitochondrial Ca2+ efflux via ΔΨm (Figs. 3 and 4).
- Overloaded Ca2+ levels have been reported in adipocytes from patients with obesity with insulin resistance [89] and from T2DM rats [90], which may link to the abnormal functions of mitochondria and ER which are unable to maintain Ca2+ homeostasis in cells. ΔΨm depolarization enhances ROS production [91-93]; however, hTFAM overexpression prevents fatty acid-induced ΔΨm depolarization in muscle cells and blocks ROS production in mouse skeletal muscle [10]. Moreover, mild ΔΨm uncoupling induced by hTFAM increases glucose uptake in skeletal muscle and improves high-fat diet-induced insulin resistance [10]. Further studies are required to determine how hTFAM regulates Ca2+ flux in mitochondria via ΔΨm.
NUCLEUS DISPATCHES TFAM TO THE MITOCHONDRIA
- This review has provided an overview of how mitochondria signaling to the nucleus and interact with the ER to regulate Ca2+ as well as the role of mitochondrial dysregulation via cellular Ca2+ homeostasis in the pathogenesis of metabolic diseases, including insulin resistance and T2DM. Although further investigation is required to determine mechanisms by which TFAM controls ΔΨm to regulate Ca2+ flux in the cell, accumulated data indicate that TFAM-driven ΔΨm regulates mitochondrial and ER’s Ca2+ flux. This leads to Ca2+ signaling into the nucleus, thereby inducing the expression of various genes such as flux channels and signaling co-factors for Ca2+. TFAM-mediated regulation of ΔΨm prevents high-fat diet-induced oxidative stress and insulin resistance via enhanced expression of GLUT4, PGC-1α, and PPARδ from the nucleus. Moreover, the mitochondria interact with the ER and regulate cellular Ca2+ flux. This process clearly influences mitochondrial TCA cycle and oxidative phosphorylation, whereas dysregulation of this process could increase metabolic diseases such as T2DM. Several fundamental cellular processes are governed by this crosstalk among the mitochondria, ER, and nucleus, which plays an essential role in the regulation of the cellular metabolic response to environmental cues. The blockage of this communication reduces mitochondrial activity [94,95] and increases the incidence rate of metabolic disease. Thus, the mitochondria govern intercommunication between cellular organelles, including ER and nucleus, via Ca2+ signaling to regulate metabolism.
- Taken together, here, we propose that TFAM is involved in the regulation of Ca2+ flux via the mitochondria-ER interaction that can signal to the nucleus, which ultimately mitigates metabolic disorders.
CONCLUSIONS
-
CONFLICTS OF INTEREST
No potential conflict of interest relevant to this article was reported.
-
FUNDING
This study was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (grant no. NRF-2019R1A2C1006334).
NOTES
-
Acknowledgements
- None


- 1. Finsterer J. Overview on visceral manifestations of mitochondrial disorders. Neth J Med 2006;64:61-71.PubMed
- 2. Patti ME, Corvera S. The role of mitochondria in the pathogenesis of type 2 diabetes. Endocr Rev 2010;31:364-95.ArticlePubMedPMC
- 3. La Rovere RM, Roest G, Bultynck G, Parys JB. Intracellular Ca(2+) signaling and Ca(2+) microdomains in the control of cell survival, apoptosis and autophagy. Cell Calcium 2016;60:74-87.ArticlePubMed
- 4. Lanner JT, Katz A, Tavi P, Sandstrom ME, Zhang SJ, Wretman C, et al. The role of Ca2+ influx for insulin-mediated glucose uptake in skeletal muscle. Diabetes 2006;55:2077-83.ArticlePubMedPDF
- 5. Ojuka EO, Jones TE, Han DH, Chen M, Wamhoff BR, Sturek M, et al. Intermittent increases in cytosolic Ca2+ stimulate mitochondrial biogenesis in muscle cells. Am J Physiol Endocrinol Metab 2002;283:E1040-5.ArticlePubMed
- 6. Rizzuto R, Brini M, Murgia M, Pozzan T. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science 1993;262:744-7.ArticlePubMed
- 7. Trost SU, Belke DD, Bluhm WF, Meyer M, Swanson E, Dillmann WH. Overexpression of the sarcoplasmic reticulum Ca(2+)-ATPase improves myocardial contractility in diabetic cardiomyopathy. Diabetes 2002;51:1166-71.ArticlePubMedPDF
- 8. Paupe V, Prudent J. New insights into the role of mitochondrial calcium homeostasis in cell migration. Biochem Biophys Res Commun 2018;500:75-86.ArticlePubMedPMC
- 9. Ojuka EO, Jones TE, Han DH, Chen M, Holloszy JO. Raising Ca2+ in L6 myotubes mimics effects of exercise on mitochondrial biogenesis in muscle. FASEB J 2003;17:675-81.ArticlePubMedPDF
- 10. Koh JH, Johnson ML, Dasari S, LeBrasseur NK, Vuckovic I, Henderson GC, et al. TFAM enhances fat oxidation and attenuates high-fat diet-induced insulin resistance in skeletal muscle. Diabetes 2019;68:1552-64.ArticlePubMedPMCPDF
- 11. Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell 2012;Mar 16 148:1145-59.ArticlePubMedPMC
- 12. Larsson NG, Barsh GS, Clayton DA. Structure and chromosomal localization of the mouse mitochondrial transcription factor A gene (Tfam). Mamm Genome 1997;8:139-40.ArticlePubMedPDF
- 13. Scarpulla RC. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev 2008;88:611-38.ArticlePubMed
- 14. Kang I, Chu CT, Kaufman BA. The mitochondrial transcription factor TFAM in neurodegeneration: emerging evidence and mechanisms. FEBS Lett 2018;592:793-811.PubMedPMC
- 15. Fisher RP, Lisowsky T, Parisi MA, Clayton DA. DNA wrapping and bending by a mitochondrial high mobility group-like transcriptional activator protein. J Biol Chem 1992;267:3358-67.ArticlePubMed
- 16. Fisher RP, Parisi MA, Clayton DA. Flexible recognition of rapidly evolving promoter sequences by mitochondrial transcription factor 1. Genes Dev 1989;3(12B):2202-17.ArticlePubMed
- 17. Larsson NG, Wang J, Wilhelmsson H, Oldfors A, Rustin P, Lewandoski M, et al. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat Genet 1998;18:231-6.ArticlePubMedPDF
- 18. Ekstrand MI, Falkenberg M, Rantanen A, Park CB, Gaspari M, Hultenby K, et al. Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum Mol Genet 2004;13:935-44.ArticlePubMed
- 19. Whelan SP, Zuckerbraun BS. Mitochondrial signaling: forwards, backwards, and in between. Oxid Med Cell Longev 2013;2013:351613.ArticlePubMedPMCPDF
- 20. Chau CM, Evans MJ, Scarpulla RC. Nuclear respiratory factor 1 activation sites in genes encoding the gamma-subunit of ATP synthase, eukaryotic initiation factor 2 alpha, and tyrosine aminotransferase. Specific interaction of purified NRF-1 with multiple target genes. J Biol Chem 1992;267:6999-7006.ArticlePubMed
- 21. Virbasius JV, Scarpulla RC. Activation of the human mitochondrial transcription factor A gene by nuclear respiratory factors: a potential regulatory link between nuclear and mitochondrial gene expression in organelle biogenesis. Proc Natl Acad Sci U S A 1994;91:1309-13.ArticlePubMedPMC
- 22. Baar K, Wende AR, Jones TE, Marison M, Nolte LA, Chen M, et al. Adaptations of skeletal muscle to exercise: rapid increase in the transcriptional coactivator PGC-1. FASEB J 2002;16:1879-86.ArticlePubMedPDF
- 23. Murakami T, Shimomura Y, Yoshimura A, Sokabe M, Fujitsuka N. Induction of nuclear respiratory factor-1 expression by an acute bout of exercise in rat muscle. Biochim Biophys Acta 1998;1381:113-22.ArticlePubMed
- 24. Piantadosi CA, Suliman HB. Mitochondrial transcription factor A induction by redox activation of nuclear respiratory factor 1. J Biol Chem 2006;281:324-33.ArticlePubMed
- 25. Miranda S, Foncea R, Guerrero J, Leighton F. Oxidative stress and upregulation of mitochondrial biogenesis genes in mitochondrial DNA-depleted HeLa cells. Biochem Biophys Res Commun 1999;258:44-9.ArticlePubMed
- 26. Suliman HB, Carraway MS, Welty-Wolf KE, Whorton AR, Piantadosi CA. Lipopolysaccharide stimulates mitochondrial biogenesis via activation of nuclear respiratory factor-1. J Biol Chem 2003;278:41510-8.ArticlePubMed
- 27. Giorgi C, Agnoletto C, Bononi A, Bonora M, De Marchi E, Marchi S, et al. Mitochondrial calcium homeostasis as potential target for mitochondrial medicine. Mitochondrion 2012;12:77-85.ArticlePubMedPMC
- 28. Denton RM, McCormack JG. On the role of the calcium transport cycle in heart and other mammalian mitochondria. FEBS Lett 1980;119:1-8.ArticlePDF
- 29. Hansford RG, Castro F. Effects of micromolar concentrations of free calcium ions on the reduction of heart mitochondrial NAD(P) by 2-oxoglutarate. Biochem J 1981;198:525-33.PubMedPMC
- 30. Murgia M, Rizzuto R. Molecular diversity and pleiotropic role of the mitochondrial calcium uniporter. Cell Calcium 2015;58:11-7.ArticlePubMed
- 31. Ainbinder A, Boncompagni S, Protasi F, Dirksen RT. Role of mitofusin-2 in mitochondrial localization and calcium uptake in skeletal muscle. Cell Calcium 2015;57:14-24.ArticlePubMed
- 32. Fujiwara Y, Kawaguchi Y, Fujimoto T, Kanayama N, Magari M, Tokumitsu H. Differential AMP-activated protein kinase (AMPK) recognition mechanism of Ca2+/calmodulin-dependent protein kinase kinase isoforms. J Biol Chem 2016;291:13802-8.ArticlePubMedPMC
- 33. Yuzefovych LV, LeDoux SP, Wilson GL, Rachek LI. Mitochondrial DNA damage via augmented oxidative stress regulates endoplasmic reticulum stress and autophagy: crosstalk, links and signaling. PLoS One 2013;8:e83349.ArticlePubMedPMC
- 34. Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol 2000;1:11-21.ArticlePubMedPDF
- 35. Raffaello A, Mammucari C, Gherardi G, Rizzuto R. Calcium at the center of cell signaling: interplay between endoplasmic reticulum, mitochondria, and lysosomes. Trends Biochem Sci 2016;41:1035-49.ArticlePubMedPMC
- 36. Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, et al. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 1998;280:1763-6.ArticlePubMed
- 37. Bach D, Naon D, Pich S, Soriano FX, Vega N, Rieusset J, et al. Expression of Mfn2, the Charcot-Marie-Tooth neuropathy type 2A gene, in human skeletal muscle: effects of type 2 diabetes, obesity, weight loss, and the regulatory role of tumor necrosis factor alpha and interleukin-6. Diabetes 2005;54:2685-93.PubMed
- 38. Sebastian D, Hernandez-Alvarez MI, Segales J, Sorianello E, Munoz JP, Sala D, et al. Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc Natl Acad Sci U S A 2012;109:5523-8.ArticlePubMedPMC
- 39. De Stefani D, Bononi A, Romagnoli A, Messina A, De Pinto V, Pinton P, et al. VDAC1 selectively transfers apoptotic Ca2+ signals to mitochondria. Cell Death Differ 2012;19:267-73.ArticlePubMedPDF
- 40. Baughman JM, Perocchi F, Girgis HS, Plovanich M, BelcherTimme CA, Sancak Y, et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011;476:341-5.ArticlePubMedPMCPDF
- 41. De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R. A fortykilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011;476:336-40.ArticlePubMedPMCPDF
- 42. Xu L, Wu Z, He Y, Chen Z, Xu K, Yu W, et al. MFN2 contributes to metabolic disorders and inflammation in the aging of rat chondrocytes and osteoarthritis. Osteoarthritis Cartilage 2020;28:1079-91.ArticlePubMed
- 43. Mitchell P, Moyle J. Chemiosmotic hypothesis of oxidative phosphorylation. Nature 1967;213:137-9.ArticlePubMedPDF
- 44. Amuthan G, Biswas G, Ananadatheerthavarada HK, Vijayasarathy C, Shephard HM, Avadhani NG. Mitochondrial stressinduced calcium signaling, phenotypic changes and invasive behavior in human lung carcinoma A549 cells. Oncogene 2002;21:7839-49.ArticlePubMedPDF
- 45. Biswas G, Adebanjo OA, Freedman BD, Anandatheerthavarada HK, Vijayasarathy C, Zaidi M, et al. Retrograde Ca2+ signaling in C2C12 skeletal myocytes in response to mitochondrial genetic and metabolic stress: a novel mode of inter-organelle crosstalk. EMBO J 1999;18:522-33.ArticlePubMedPMC
- 46. Watanabe A, Arai M, Koitabashi N, Niwano K, Ohyama Y, Yamada Y, et al. Mitochondrial transcription factors TFAM and TFB2M regulate Serca2 gene transcription. Cardiovasc Res 2011;90:57-67.ArticlePubMed
- 47. Aydin J, Andersson DC, Hanninen SL, Wredenberg A, Tavi P, Park CB, et al. Increased mitochondrial Ca2+ and decreased sarcoplasmic reticulum Ca2+ in mitochondrial myopathy. Hum Mol Genet 2009;18:278-88.ArticlePubMed
- 48. Cutler MJ, Wan X, Plummer BN, Liu H, Deschenes I, Laurita KR, et al. Targeted sarcoplasmic reticulum Ca2+ ATPase 2a gene delivery to restore electrical stability in the failing heart. Circulation 2012;126:2095-104.ArticlePubMedPMC
- 49. Sommakia S, Houlihan PR, Deane SS, Simcox JA, Torres NS, Jeong MY, et al. Mitochondrial cardiomyopathies feature increased uptake and diminished efflux of mitochondrial calcium. J Mol Cell Cardiol 2017;113:22-32.ArticlePubMedPMC
- 50. Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature 2004;427:360-4.ArticlePubMedPDF
- 51. Samanta K, Mirams GR, Parekh AB. Sequential forward and reverse transport of the Na+ Ca2+ exchanger generates Ca2+ oscillations within mitochondria. Nat Commun 2018;9:156.ArticlePubMedPMCPDF
- 52. Jornayvaz FR, Shulman GI. Regulation of mitochondrial biogenesis. Essays Biochem 2010;47:69-84.ArticlePubMedPDF
- 53. Poyton RO, McEwen JE. Crosstalk between nuclear and mitochondrial genomes. Annu Rev Biochem 1996;65:563-607.ArticlePubMed
- 54. Fischer C, DeFalco TA, Karia P, Snedden WA, Moeder W, Yoshioka K, et al. Calmodulin as a Ca2+-sensing subunit of Arabidopsis cyclic nucleotide-gated channel complexes. Plant Cell Physiol 2017;58:1208-21.ArticlePubMed
- 55. Rose AJ, Kiens B, Richter EA. Ca2+-calmodulin-dependent protein kinase expression and signalling in skeletal muscle during exercise. J Physiol 2006;574(Pt 3):889-903.ArticlePubMedPMC
- 56. Jager S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci U S A 2007;104:12017-22.PubMedPMC
- 57. Koh JH, Hancock CR, Han DH, Holloszy JO, Nair KS, Dasari S. AMPK and PPARβ positive feedback loop regulates endurance exercise training-mediated GLUT4 expression in skeletal muscle. Am J Physiol Endocrinol Metab 2019;316:E931-9.ArticlePubMedPMC
- 58. Rowland AA, Voeltz GK. Endoplasmic reticulum-mitochondria contacts: function of the junction. Nat Rev Mol Cell Biol 2012;13:607-25.ArticlePubMedPMCPDF
- 59. Yang C, Watson RT, Elmendorf JS, Sacks DB, Pessin JE. Calmodulin antagonists inhibit insulin-stimulated GLUT4 (glucose transporter 4) translocation by preventing the formation of phosphatidylinositol 3,4,5-trisphosphate in 3T3L1 adipocytes. Mol Endocrinol 2000;14:317-26.ArticlePubMed
- 60. Contreras-Ferrat AE, Toro B, Bravo R, Parra V, Vasquez C, Ibarra C, et al. An inositol 1,4,5-triphosphate (IP3)-IP3 receptor pathway is required for insulin-stimulated glucose transporter 4 translocation and glucose uptake in cardiomyocytes. Endocrinology 2010;151:4665-77.ArticlePubMedPDF
- 61. Vial G, Dubouchaud H, Couturier K, Cottet-Rousselle C, Taleux N, Athias A, et al. Effects of a high-fat diet on energy metabolism and ROS production in rat liver. J Hepatol 2011;54:348-56.ArticlePubMed
- 62. Fu S, Yang L, Li P, Hofmann O, Dicker L, Hide W, et al. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 2011;473:528-31.ArticlePubMedPMCPDF
- 63. Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004;306:457-61.ArticlePubMed
- 64. Tubbs E, Theurey P, Vial G, Bendridi N, Bravard A, Chauvin MA, et al. Mitochondria-associated endoplasmic reticulum membrane (MAM) integrity is required for insulin signaling and is implicated in hepatic insulin resistance. Diabetes 2014;63:3279-94.ArticlePubMedPDF
- 65. Marchi S, Marinello M, Bononi A, Bonora M, Giorgi C, Rimessi A, et al. Selective modulation of subtype III IP₃R by Akt regulates ER Ca²+ release and apoptosis. Cell Death Dis 2012;3:e304.ArticlePubMedPMCPDF
- 66. Szado T, Vanderheyden V, Parys JB, De Smedt H, Rietdorf K, Kotelevets L, et al. Phosphorylation of inositol 1,4,5-trisphosphate receptors by protein kinase B/Akt inhibits Ca2+ release and apoptosis. Proc Natl Acad Sci U S A 2008;105:2427-32.ArticlePubMedPMC
- 67. Murley A, Lackner LL, Osman C, West M, Voeltz GK, Walter P, et al. ER-associated mitochondrial division links the distribution of mitochondria and mitochondrial DNA in yeast. Elife 2013;2:e00422.ArticlePubMedPMCPDF
- 68. Lewis SC, Uchiyama LF, Nunnari J. ER-mitochondria contacts couple mtDNA synthesis with mitochondrial division in human cells. Science 2016;353:aaf5549.ArticlePubMedPMC
- 69. Chen H, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM, et al. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 2010;141:280-9.ArticlePubMedPMC
- 70. Gan KX, Wang C, Chen JH, Zhu CJ, Song GY. Mitofusin-2 ameliorates high-fat diet-induced insulin resistance in liver of rats. World J Gastroenterol 2013;19:1572-81.ArticlePubMedPMC
- 71. Chitra L, Boopathy R. Altered mitochondrial biogenesis and its fusion gene expression is involved in the high-altitude adaptation of rat lung. Respir Physiol Neurobiol 2014;192:74-84.ArticlePubMed
- 72. Alam TI, Kanki T, Muta T, Ukaji K, Abe Y, Nakayama H, et al. Human mitochondrial DNA is packaged with TFAM. Nucleic Acids Res 2003;31:1640-5.ArticlePubMedPMC
- 73. Kaufman BA, Durisic N, Mativetsky JM, Costantino S, Hancock MA, Grutter P, et al. The mitochondrial transcription factor TFAM coordinates the assembly of multiple DNA molecules into nucleoid-like structures. Mol Biol Cell 2007;18:3225-36.ArticlePubMedPMC
- 74. Ngo HB, Kaiser JT, Chan DC. The mitochondrial transcription and packaging factor Tfam imposes a U-turn on mitochondrial DNA. Nat Struct Mol Biol 2011;18:1290-6.ArticlePubMedPMCPDF
- 75. Belke DD, Swanson E, Suarez J, Scott BT, Stenbit AE, Dillmann WH. Increased expression of SERCA in the hearts of transgenic mice results in increased oxidation of glucose. Am J Physiol Heart Circ Physiol 2007;292:H1755-63.ArticlePubMed
- 76. Suarez J, Scott B, Dillmann WH. Conditional increase in SERCA2a protein is able to reverse contractile dysfunction and abnormal calcium flux in established diabetic cardiomyopathy. Am J Physiol Regul Integr Comp Physiol 2008;295:R1439-45.ArticlePubMedPMC
- 77. Kunkel GH, Chaturvedi P, Tyagi SC. Mitochondrial pathways to cardiac recovery: TFAM. Heart Fail Rev 2016;21:499-517.ArticlePubMedPMCPDF
- 78. Neeb ZP, Edwards JM, Alloosh M, Long X, Mokelke EA, Sturek M. Metabolic syndrome and coronary artery disease in Ossabaw compared with Yucatan swine. Comp Med 2010;60:300-15.PubMedPMC
- 79. Perocchi F, Gohil VM, Girgis HS, Bao XR, McCombs JE, Palmer AE, et al. MICU1 encodes a mitochondrial EF hand protein required for Ca(2+) uptake. Nature 2010;467:291-6.ArticlePubMedPMCPDF
- 80. Plovanich M, Bogorad RL, Sancak Y, Kamer KJ, Strittmatter L, Li AA, et al. MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS One 2013;8:e55785.ArticlePubMedPMC
- 81. Raffaello A, De Stefani D, Sabbadin D, Teardo E, Merli G, Picard A, et al. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J 2013;32:2362-76.ArticlePubMedPMC
- 82. Sancak Y, Markhard AL, Kitami T, Kovacs-Bogdan E, Kamer KJ, Udeshi ND, et al. EMRE is an essential component of the mitochondrial calcium uniporter complex. Science 2013;342:1379-82.ArticlePubMedPMC
- 83. Vecellio Reane D, Vallese F, Checchetto V, Acquasaliente L, Butera G, De Filippis V, et al. A MICU1 splice variant confers high sensitivity to the mitochondrial Ca2+ uptake machinery of skeletal muscle. Mol Cell 2016;64:760-73.ArticlePubMed
- 84. Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol 2013;15:1464-72.ArticlePubMedPMCPDF
- 85. Gherardi G, Nogara L, Ciciliot S, Fadini GP, Blaauw B, Braghetta P, et al. Loss of mitochondrial calcium uniporter rewires skeletal muscle metabolism and substrate preference. Cell Death Differ 2019;26:362-81.ArticlePubMedPDF
- 86. Koves TR, Ussher JR, Noland RC, Slentz D, Mosedale M, Ilkayeva O, et al. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab 2008;7:45-56.ArticlePubMed
- 87. Hattori Y, Matsuda N, Kimura J, Ishitani T, Tamada A, Gando S, et al. Diminished function and expression of the cardiac Na+- Ca2+ exchanger in diabetic rats: implication in Ca2+ overload. J Physiol 2000;527(Pt 1):85-94.ArticlePubMedPMCPDF
- 88. Kashihara H, Shi ZQ, Yu JZ, McNeill JH, Tibbits GF. Effects of diabetes and hypertension on myocardial Na+-Ca2+ exchange. Can J Physiol Pharmacol 2000;78:12-9.ArticlePubMed
- 89. Draznin B, Sussman KE, Eckel RH, Kao M, Yost T, Sherman NA. Possible role of cytosolic free calcium concentrations in mediating insulin resistance of obesity and hyperinsulinemia. J Clin Invest 1988;82:1848-52.ArticlePubMedPMC
- 90. Draznin B, Lewis D, Houlder N, Sherman N, Adamo M, Garvey WT, et al. Mechanism of insulin resistance induced by sustained levels of cytosolic free calcium in rat adipocytes. Endocrinology 1989;125:2341-9.ArticlePubMed
- 91. Barros MH, Bandy B, Tahara EB, Kowaltowski AJ. Higher respiratory activity decreases mitochondrial reactive oxygen release and increases life span in Saccharomyces cerevisiae. J Biol Chem 2004;279:49883-8.ArticlePubMed
- 92. Davidson JF, Schiestl RH. Mitochondrial respiratory electron carriers are involved in oxidative stress during heat stress in Saccharomyces cerevisiae. Mol Cell Biol 2001;21:8483-9.ArticlePubMedPMCPDF
- 93. Gourlay CW, Carpp LN, Timpson P, Winder SJ, Ayscough KR. A role for the actin cytoskeleton in cell death and aging in yeast. J Cell Biol 2004;164:803-9.ArticlePubMedPMCPDF
- 94. Barrientos A, Moraes CT. Simultaneous transfer of mitochondrial DNA and single chromosomes in somatic cells: a novel approach for the study of defects in nuclear-mitochondrial communication. Hum Mol Genet 1998;7:1801-8.ArticlePubMed
- 95. Mastromonaco GF, Favetta LA, Smith LC, Filion F, King WA. The influence of nuclear content on developmental competence of gaur x cattle hybrid in vitro fertilized and somatic cell nuclear transfer embryos. Biol Reprod 2007;76:514-23.PubMed
REFERENCES
Figure & Data
References
Citations
- Targeted metabolomics reveals the aberrant energy status in diabetic peripheral neuropathy and the neuroprotective mechanism of traditional Chinese medicine JinMaiTong
Bingjia Zhao, Qian Zhang, Yiqian He, Weifang Cao, Wei Song, Xiaochun Liang
Journal of Pharmaceutical Analysis.2024; 14(2): 225. CrossRef - Mitochondrial damage‐associated molecular patterns: A new insight into metabolic inflammation in type 2 diabetes mellitus
Yan Wang, Jingwu Wang, Si‐Yu Tao, Zhengting Liang, Rong xie, Nan‐nan Liu, Ruxue Deng, Yuelin Zhang, Deqiang Deng, Guangjian Jiang
Diabetes/Metabolism Research and Reviews.2024;[Epub] CrossRef - Altered Energy Metabolism, Mitochondrial Dysfunction, and Redox Imbalance Influencing Reproductive Performance in Granulosa Cells and Oocyte During Aging
Hiroshi Kobayashi, Chiharu Yoshimoto, Sho Matsubara, Hiroshi Shigetomi, Shogo Imanaka
Reproductive Sciences.2024; 31(4): 906. CrossRef - When Our Best Friend Becomes Our Worst Enemy: The Mitochondrion in Trauma, Surgery, and Critical Illness
May-Kristin Torp, Kåre-Olav Stensløkken, Jarle Vaage
Journal of Intensive Care Medicine.2024;[Epub] CrossRef - Attenuating mitochondrial dysfunction and morphological disruption with PT320 delays dopamine degeneration in MitoPark mice
Vicki Wang, Kuan-Yin Tseng, Tung-Tai Kuo, Eagle Yi-Kung Huang, Kuo-Lun Lan, Zi-Rong Chen, Kuo-Hsing Ma, Nigel H. Greig, Jin Jung, Ho-II Choi, Lars Olson, Barry J. Hoffer, Yuan-Hao Chen
Journal of Biomedical Science.2024;[Epub] CrossRef - Effects of the anti-inflammatory drug celecoxib on cell death signaling in human colon cancer
Ryuto Maruyama, Yuki Kiyohara, Yasuhiro Kudo, Tomoyasu Sugiyama
Naunyn-Schmiedeberg's Archives of Pharmacology.2023; 396(6): 1171. CrossRef - gp130 Activates Mitochondrial Dynamics for Hepatocyte Survival in a Model of Steatohepatitis
Daria Shunkina, Anastasia Dakhnevich, Egor Shunkin, Olga Khaziakhmatova, Valeria Shupletsova, Maria Vulf, Alexandra Komar, Elena Kirienkova, Larisa Litvinova
Biomedicines.2023; 11(2): 396. CrossRef - Pharmacological Activation of Rev-erbα Attenuates Doxorubicin-Induced Cardiotoxicity by PGC-1α Signaling Pathway
Runmei Zou, Shuo Wang, Hong Cai, Yuwen Wang, Cheng Wang, Vivek Pandey
Cardiovascular Therapeutics.2023; 2023: 1. CrossRef - Protective Effect of Ergothioneine against 7-Ketocholesterol-Induced Mitochondrial Damage in hCMEC/D3 Human Brain Endothelial Cells
Damien Meng-Kiat Leow, Irwin Kee-Mun Cheah, Zachary Wei-Jie Fong, Barry Halliwell, Wei-Yi Ong
International Journal of Molecular Sciences.2023; 24(6): 5498. CrossRef - Effect of PPARγ on oxidative stress in diabetes-related dry eye
Jing Wang, Shuangping Chen, Xiuxiu Zhao, Qian Guo, Ruibo Yang, Chen Zhang, Yue Huang, Lechong Ma, Shaozhen Zhao
Experimental Eye Research.2023; 231: 109498. CrossRef - Chiisanoside Mediates the Parkin/ZNF746/PGC-1α Axis by Downregulating MiR-181a to Improve Mitochondrial Biogenesis in 6-OHDA-Caused Neurotoxicity Models In Vitro and In Vivo: Suggestions for Prevention of Parkinson’s Disease
Yu-Ling Hsu, Hui-Jye Chen, Jia-Xin Gao, Ming-Yang Yang, Ru-Huei Fu
Antioxidants.2023; 12(9): 1782. CrossRef - TBBPA causes apoptosis in grass carp hepatocytes involving destroyed ER-mitochondrial function
Dongxu Han, Naixi Yang, Huanyi Liu, Yujie Yao, Shiwen Xu
Chemosphere.2023; 341: 139974. CrossRef - The Protective Mechanism of TFAM on Mitochondrial DNA and its Role in Neurodegenerative Diseases
Ying Song, Wenjun Wang, Beibei Wang, Qiwen Shi
Molecular Neurobiology.2023;[Epub] CrossRef - Impact of Roux-en-Y Gastric Bypass on Mitochondrial Biogenesis and Dynamics in Leukocytes of Obese Women
Zaida Abad-Jiménez, Teresa Vezza, Sandra López-Domènech, Meylin Fernández-Reyes, Francisco Canet, Carlos Morillas, Segundo Ángel Gómez-Abril, Celia Bañuls, Víctor M. Víctor, Milagros Rocha
Antioxidants.2022; 11(7): 1302. CrossRef - The Effects of Galgunhwanggumhwangryun-tang on Glucose and Energy Metabolism in C2C12 Myotubes
Jihong Oh, Song-Yi Han, Soo Kyoung Lim, Hojun Kim
Journal of Korean Medicine for Obesity Research.2022; 22(2): 93. CrossRef
 PubReader
PubReader ePub Link
ePub Link Cite
Cite