- Current
- Browse
- Collections
-
For contributors
- For Authors
- Instructions to authors
- Article processing charge
- e-submission
- For Reviewers
- Instructions for reviewers
- How to become a reviewer
- Best reviewers
- For Readers
- Readership
- Subscription
- Permission guidelines
- About
- Editorial policy
Articles
- Page Path
- HOME > Diabetes Metab J > Volume 35(3); 2011 > Article
-
Sulwon Lecture 2010Mitochondrial Dysfunction and Insulin Resistance: The Contribution of Dioxin-Like Substances
- Hong Kyu Lee
-
Diabetes & Metabolism Journal 2011;35(3):207-215.
DOI: https://doi.org/10.4093/dmj.2011.35.3.207
Published online: June 30, 2011
Department of Internal Medicine, Eulji University School of Medicine, Seoul, Korea.
- Corresponding author: Hong Kyu Lee. Department of Internal Medicine, Eulji Hospital, 280-1 Hagye 1-dong, Nowon-gu, Seoul 139-711, Korea. hkleemd@eulji.ac.kr
Copyright © 2011 Korean Diabetes Association
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- ABSTRACT
- INTRODUCTION TO INSULIN RESISTANCE AND THE MITOCHONDRIA-BASED MODEL OF METABOLIC SYNDROME
- MITOCHONDRIA-BASED MODEL OF THE METABOLIC SYNDROME: A SPECIAL CASE OF METABOLIC SCALING
- CAUSES OF TYPE 2 DIABETES
- ESTABLISHMENT OF POPS AS A CAUSE OF DIABETES
- PROBLEMS IN ESTABLISHING A CAUSE AND EFFECT RELATIONSHIP BETWEEN POP EXPOSURE AND DEVELOPMENT OF THE METABOLIC SYNDROME
- BIOASSAY OF SERUM DIOXIN-LIKE ACTIVITY, A BREAKTHROUGH?
- CRITIQUES OF THE MITOCHONDRIAL HYPOTHESIS
- SUMMARY AND CONCLUSIONS
- ACKNOWLEDGMENTS
- NOTES
- REFERENCES
ABSTRACT
- Persistent organic pollutants (POPs) are known to cause mitochondrial dysfunction and this in turn is linked to insulin resistance, a key biochemical abnormality underlying the metabolic syndrome. To establish the cause and effect relationship between exposure to POPs and the development of the metabolic syndrome, Koch's postulates were considered. Problems arising from this approach were discussed and possible solutions were suggested. In particular, the difficulty of establishing a cause and effect relationship due to the vagueness of the metabolic syndrome as a disease entity was discussed. Recently a bioassay, aryl-hydrocarbon receptor (AhR) trans-activation activity using a cell line expressing AhR-luciferase, showed that its activity is linearly related with the parameters of the metabolic syndrome in a population. This finding suggests the possible role of bioassays in the analysis of multiple pollutants of similar kinds in the pathogenesis of several closely related diseases, such as type 2 diabetes and the metabolic syndrome. Understanding the effects of POPs on mitochondrial function will be very useful in understanding the integration of various factors involved in this process, such as genes, fetal malnutrition and environmental toxins and their protectors, as mitochondria act as a unit according to the metabolic scaling law.
- Insulin resistance is a common biochemical abnormality preceding type 2 diabetes and other associated diseases. In 1988, G. Reaven proposed that this state should be studied as a disease and named it syndrome X, or unknown syndrome. Epidemiologic studies revealed the risk factors of this disease and ultimately the cause(s). The World Health Organization (WHO) gave a new name to this disease, metabolic syndrome, and established its diagnostic criteria in 1998. The metabolic syndrome is defined as the presence of at least 3 of the following symptoms or phenotypes in an individual; abdominal obesity (measured by waist circumference), high serum triglyceride level, low serum high density lipoprotein (HDL) level, hyperglycemia, and high blood pressure.
- In 1998, my colleagues and I found that blood mitochondrial DNA (mtDNA) density in peripheral blood cells was lower in those subjects who would go on to develop diabetes in 2 years, in comparison to those who would not. Furthermore, people with lower blood mtDNA density had high blood pressure, high serum triglyceride levels and a high waist circumference ratio. These findings suggest that a decrease in mtDNA density in blood cells is causally related to the metabolic syndrome. In the following years, we accumulated evidence suggesting that mitochondrial dysfunction might be the central abnormality causing insulin resistance, and it might also be responsible for the insulin secretion defect found with type 2 diabetes. We presented evidence in support of this hypothesis and this evidence underwent several reviews [1,2].
- In this lecture, I will argue again that the metabolic syndrome is a state of mitochondrial dysfunction, which is, in turn, identical to the state of insulin resistance. In this interpretation, cause(s) of type 2 diabetes are identical to cause(s) of the metabolic syndrome, and type 2 diabetes is later stage of this altered body state. The insulin secretion defect in type 2 diabetes might arise from the insulin resistance state per se, as it is a state of mitochondrial dysfunction and good mitochondrial function is needed for good insulin secretion. This view of the metabolic syndrome is thus narrower than the generally held view, which includes full-blown type 2 diabetes and atherosclerosis within the metabolic syndrome (Fig. 1).
INTRODUCTION TO INSULIN RESISTANCE AND THE MITOCHONDRIA-BASED MODEL OF METABOLIC SYNDROME
- Another important aspect of the mitochondria-based model is that it is a special case of the scaling law. We know that many body parameters correlate with body size; the bigger the body size, the larger the metabolism rate becomes. It is well known that the metabolic rate of mammals increases when body mass increases. There is a linear relationship between metabolic rate (Y), and body mass (M) characterized by the equation, Y= Y0×M(body mass)3/4.
- This law tells us that the larger the body size, the greater the decrease in the mass specific metabolic rate (Y/M). Most importantly, it was found that this relationship holds down to individual cells and their mitochondria as well as mitochondrial electron transfer chain activities [3]. In other words, if the body size of a mammal increases, its unit (cell or mitochondrial) metabolic power decreases (Fig. 2). We found that mitochondrial DNA copy numbers are decreased in the metabolic syndrome, and this is negatively correlated with serum insulin responses in health young male medical students. From these results, we reasoned that the metabolic syndrome is the result of the metabolic scaling law in action, in that body metabolism is changing in order to adapt to decreasing mitochondrial function. In other words, the state of insulin resistance is an adaptation of the body to the decrease in unit mitochondrial function. This reasoning is also discussed in detail in a previous paper [2].
- One of the key principles of this process is the adaptation of enzymes to the body temperature of 37℃. At this temperature, most enzymes have the lowest Gibbs free energy. If mitochondrial dysfunction leads to a decrease in heat production, body temperature would be lower, and this has to be compensated for by an increase in fat mass (insulation), increased heat production, and thus more mitochondrial mass and nutrient supply, which in turn would require higher levels of serum insulin, glucose, and triglycerides.
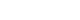
- Metabolic scaling is the only "law" generally accepted in biology. The term "law" is not debated in this usage. Scaling is applicable not only to the metabolic rate but to many aspects of body states, such as the relationship between body size and brain mass, aorta circumference, blood volume and almost every aspect of body metabolism. This phenomenon is also known as "allometry"; where the size of every part of body is quantitatively related to the whole (mass). It is not surprising that bone mass and bone strength should increase along with the body size of an animal; lung volume should increase with increasing metabolic demand, kidney size also increases in order to deal with the increasing metabolic waste of an increasing body mass and blood volume. This concept is illustrated in Fig. 3.
- This kind of relationship predicts that if body mass increases (obesity), the circulatory system will enlarge in adaptation. However, there will be limit to this adaptation, in particular when unit cell (or mitochondrial function) is decreased, as in the insulin resistance state, a mismatch will develop. If heart metabolism (i.e., cardiac output) did not increase in order to adapt to increasing demand from the whole body, the result would be heart failure. If the coronary artery does not need to supply as much blood to the heart due to a decrease in energy demand, the coronary artery should decrease in size. I suspect that this may be a reason why coronary heart disease develops in type 2 diabetes.
- These novel explanations might be unfamiliar to most clinicians, as they are biophysical and based mostly on bioenergetics. While many aspects of this theory need experimental support, I believe that a mitochondria-based model fits well with reality.
MITOCHONDRIA-BASED MODEL OF THE METABOLIC SYNDROME: A SPECIAL CASE OF METABOLIC SCALING
- There are many "causes" in medicine; in fact "causes" are diverse in different disease states, and diseases are themselves diverse and variously defined. These features are illustrated in Fig. 1. Diabetologists may regard coronary heart disease as a complication of type 2 diabetes, while cardiologists might consider diabetes to be a cause of coronary heart disease. This difference arises because specialists treat a limited group of people with specific diseases, such as patients with type 2 diabetes or coronary heart disease. They examine why their patients get those illnesses (causes) and what might happen to them as a result (complications).
- Causes are divided into genetic and environmental causes, which can in turn be divided again into their individual components. Medicine adopts the division of "causes" when there are different treatments or prevention methods available for these different causes. For example, diabetes is caused by insulin deficiency and it is treated with an insulin supply. If you were to ask whether we could restore insulin deficiency, the answer is no. If you were to further ask why we could not do this, there would be another answer(s); such as ageing, etc. In fact, we do not know.
- We are, however, fairly certain of all of these chains of events from the genetic make-up (factors) and epigenetic changes which occur during development, particularly during the fetal stage, such as malnutrition and environmental factors. In the mitochondria-based model, there are 3 basic causes; genes and developmental and environmental toxins (and protectors). I will explain why the continuous accumulation of environmental toxins or so called persistent organic pollutants (POPs) in our body might be one of the most important factors.
- Genetic causes of type 2 diabetes; contribution of the mitochondrial genome
- Humans and other animals have two genetic systems; a nuclear genome and a mitochondrial genome. The contribution of nuclear genes to type 2 diabetes is well known and will not be discussed here. However, mtDNA, the second genome, needs attention. It is now well established that certain mtDNA polymorphism and haplotypes are either susceptible (16189 T>C, haplotype B and F) or resistant (haplotype N9a) to type 2 diabetes among Asians [5,6]. Readers are reminded that these findings were not confirmed in Europe and most European geneticists do not consider mtDNA in their study of type 2 diabetes, although a recent study in Italy showed that the 16189 T>C variant is associated with the metabolic syndrome [7].
- These facts describe the essential part of the mitochondria-based model. This model suggests that an analysis of the genetic effect(s) of nuclear genes(s) should consider the independent effects of the mitochondrial genome, at least in Asia.
- I am well aware that mitochondrial dysfunction does not cause the metabolic syndrome in general. Children born with mitochondrial DNA mutations manifest various clinical syndromes, such as mitochondrial encephalopathy, lactic acidosis and stroke-like episodes (MELAS), myoclonic epilepsy with ragged red fibers (MERRF), and Lebers hereditary optic neuropathy (LHON). These disease states are associated with a low body mass, not obesity. However, a family with a special mtDNA mutation exhibited hypertension and hyperlipidemia, suggesting that the metabolic syndrome might result from a mtDNA abnormality [8].
- Malnutrition during fetal and early life; thrifty phenotype and mitochondrial state
- Exposure to malnutrition early in life predisposes an individual to type 2 diabetes and several chronic diseases, including the metabolic syndrome. As shown in Fig. 1, if the mitochondria-based model is correct, fetal malnutrition would induce an altered mitochondrial state. We have proved this in an animal experiment [9].
- Environmental toxins; POPs
- The most important cause of type 2 diabetes and the metabolic syndrome is environmental. Something bad was introduced during industrialization and is increasing in the environment. We know that it is noninfectious; the usual suspects are "POPs." POPs are defined as "chemical substances that persist in the environment, bio-accumulate through the food web, and pose a risk of causing adverse effects to human health and the environment" by the Stockholm Convention. It identified 12 POPs initially and subsequently added 9 more.
- Pesticides: aldrin, chlordane, DDT, dieldrin, endrin, heptachlor, hexachlorobenzene, mirex, toxaphene, chlordecone, alpha hexachlorocyclohexane, beta hexachlorocyclohexane, lindane, pentachlorobenzene
- Industrial chemicals: hexachlorobenzene, polychlorinated biphenyls (PCBs), hexabromobiphenyl, hexabromodiphenyl ether and heptabromodiphenyl ether, pentachlorobenzene, perfluorooctane sulfonic acid, its salts, and perfluorooctane sulfonyl fluoride, tetrabromodiphenyl ether and pentabromodiphenyl ether
- Their by-products: hexachlorobenzene; polychlorinated dibenzo-p-dioxins and polychlorinated dibenzofurans (PCDD/PCDF), and PCBs, alpha hexachlorocyclohexane, beta hexachlorocyclohexane and pentachlorobenzene.
- (Source: http://chm.pops.int/Convention/tabid/54/language/en-US/Default.aspx.)
CAUSES OF TYPE 2 DIABETES
- Association studies
- These usual suspects were first examined by Baillie-Hamilton [10], in 2002 . She wondered if the industrial use of new chemicals after World War II might be responsible for the obesity epidemic, simply because their appearances coincided. They have been firmly indicted by a Korean epidemiologist, Lee and her colleagues [11,12], who confirmed the association between serum POPs levels and type 2 diabetes and the metabolic syndrome.
- Using the NHANES 1999-2000 data set, she found six POPs (2,2,4,4,5,5-hexachlorobiphenyl [PCB153], 1,2,3,4,6,7,8-heptachlorodibenzo-p-dioxin [HpCDD], 1,2,3,4,6,7,8,9-octachlorodibenzo-p-dioxin [OCDD], oxychlordane, p,p-dichlorodiphenyltrichloroethane [DDE], and trans-nonachlor) that were detected in at least 80% of study subjects and body load of persistent organic pollutants (sum of 6 POPs) was associated with the prevalence of diabetes in a strong dose-dependent manner. Observations showing an association between POPs and diabetes soon followed from Denmark (Greenland Inuit), Taiwan, Japan, the Slovak Republic and many other countries. These data appear too strong to deny a causal association. However, the mere presence of a statistical association between two variables, POPs and the diabetes/metabolic syndrome in our case, does not establish a cause and effect relationship.
- It is important to note that several prospective studies of selected POPs performed in occupational or accidental high exposure settings reported inconsistent results, particularly for 2,3,7,8-TCDD. Also, the decreasing trend of organochlorine POPs during recent decades is inconsistent with the current trend towards an increased prevalence of type 2 diabetes [13].
- Animal studies
- In order to establish the cause and effect relationship between POP exposure and development of the diabetes/metabolic syndrome, experimental studies are needed. We have paid particular attention to the herbicide atrazine and related triazine herbicides because they have been extensively used in the USA since the early 1960s, which corresponds to the beginning of the present obesity epidemic and they are also known to inhibit tissue respiration in mussels. Atrazine is routinely found as a contaminant in surface and ground water in the USA. It is known to bind irreversibly to the photosystem II complex on the thylakoid membranes in chloroplasts, an essential component of photosynthesis, and we found that it inhibits the electron transport of mitochondria as well. We fed rats with atrazine-containing water and found that they develop insulin resistance, a fatty liver and abdominal obesity [14].
- Recently, Ruzzin et al. [15] exposed Wistar rats to lipophilic POPs for 28 days by feeding them a high-fat diet containing crude fish oil obtained from farmed Atlantic salmon, which are contaminated with several organic pollutants. They measured body weight, whole-body insulin sensitivity, POP accumulation, and the lipid and glucose homeostasis of contaminated-salmon-oil-fed rats and compared measurements in rats fed decontaminated (with activated charcoal) salmon oil. Rats exposed to crude, but not refined, salmon oil developed insulin resistance, abdominal obesity, and hepato-steatosis. They concluded that exposure to POPs commonly present in the food chain leads to insulin resistance and associated metabolic disorders. These results are very convincing, but human studies are needed in order to unequivocally prove that POP exposure is a cause of the diabetes/metabolic syndrome.
- Nested case control study
- We cannot conduct this feeding experiment in humans. In order to establish a cause and effect relationship in humans, Lee DH approached the Coronary Artery Risk Development in Young Adults (CARDIA) study group and measured POP levels in their population before the development of disease phenotypes. She reported that serum levels of some POPs (in particular, trans-nonachlor and highly chlorinated PCBs) at CARDIA year 2 were associated with the incidence of type 2 diabetes over the next 18 years, especially in obese persons [13]. Parallel to the prediction of type 2 diabetes, many statistically significant associations of POPs with metabolic conditions appeared at low doses, forming inverted U-shaped dose-response relationships.
- Among organochlorine pesticides, the presence of p,p'-DDE predicted higher body mass index (BMI), triglycerides, and homeostasis model assessment of insulin resistance (HOMA-IR) and lower HDL-cholesterol (HDL-C) most consistently at year 20 after adjusting for baseline values. The presence of oxychlordane, trans-nonachlor, and hexachlorobenzene also significantly predicted higher triglycerides. Persistent PCBs with ≥7 chlorides predicted higher BMI, triglycerides, and HOMA-IR and lower HDL-C at year 20 with similar dose-response curves. Simultaneous exposure to various POPs in the general population appeared to contribute to the development of obesity, dyslipidemia, and insulin resistance. The authors concluded that POP exposure may contribute to excess adiposity and other features of dysmetabolism [16].
- Because POPs are well-known endocrine disruptors, they reasoned that serum POP levels showed strong associations at relatively low exposures, thus an inverted U-shape relationship characterizes this association. Unlike the traditional paradigm of cellular toxicity in which a linear dose-response relationship is the rule, the inverted U-shaped responses of endocrine disruptors have been well known among toxicologists.
ESTABLISHMENT OF POPS AS A CAUSE OF DIABETES
- A cause and effect relationship between a microorganism and disease is usually established when Koch's postulates are met; 1) the microorganism must be found in abundance in all organisms suffering from the disease, but should not be found in healthy organisms; 2) The microorganism must be isolated from a diseased organism and grown in pure culture; 3) The cultured microorganism should cause disease when introduced into a healthy organism; and 4) the microorganism must be re-isolated from the inoculated, diseased experimental host and identified as being identical to the original specific causative agent. The evidence provided above would be sufficient enough to meet Koch's postulates. As POPs are inorganic, very stable and persistent, the issues of isolation for POPs and pure culture as required in postulates 2 and 4 are irrelevant.
- We are dealing with a novel problem here; the suspected causative agents, POPs, are not a single agent, but are diverse, very similar to one another, interact with each other (variously acting as antagonist or agonist), and exhibit a "low dose effect." How can we "know" if they cause a disease, when they accumulate over many years and exhibit such complicated behaviors? There is another problem as well, because the metabolic syndrome is not a well-defined disease entity. Lee DH found that serum levels of organochlorine pesticides are more closely associated with type 2 diabetes, and abdominal obesity and high serum triglyceride levels and furans and dioxins are more closely associated with hypertension. Should we divide the metabolic syndrome into two subgroups: type 1, organochlorine pesticide syndrome and type 2, furan/dioxin syndrome? Furthermore, there are 5 or more phenotypes characterizing the metabolic syndrome and only 3 of them are needed for a diagnosis. What would be the merit of diagnosing a disease, such as the metabolic syndrome, if serum POP levels (or burden of body POPs) is high? We need to reduce POP levels anyway, regardless of the subject's phenotype. It might be worthwhile to note that the NHANES data showed that POP levels interact with BMI in determining type 2 diabetes. When POP levels were lower, diabetes prevalence was not associated with BMI, while obese people with high serum POP levels had an increase in diabetes prevalence.
PROBLEMS IN ESTABLISHING A CAUSE AND EFFECT RELATIONSHIP BETWEEN POP EXPOSURE AND DEVELOPMENT OF THE METABOLIC SYNDROME
- Measuring exposure to POPs in humans is particularly difficult as the current method of POP detection usually requires a large serum sample. There is no stored serum sample large enough to test this concept epidemiologically, only a few milliliters at best. Furthermore, measuring POPs chemically with the use of high resolution GC/MS, which is the gold standard, is very difficult, prohibitively expensive and requires a machine devoted to this use, facility and people.
- We wondered if there would be an alternative method for measuring the body burden of POPs and found the CALUX assay promising. This assay has been successfully used by environmental scientists for the screening of dioxin-like substances for many years. It takes advantage of the fact that dioxin and dioxin-like substances act through aryl-hydrocarbon receptors almost exclusively. If one were to make a cell line transfected with the plasmid expressing aryl-hydrocarbon receptor (AhR) gene, it would respond to dioxin or dioxin-like chemical substances. If AhR is Chemical (dioxin-like substances) Activated and Luciferase Expression (CALUX) is measured, one could calculate the amount of dioxin or dioxin-like substances in samples.
- Recently, KimPak Y at Kyung Hee University developed a modified CALUX assay and tested sera of pre-diabetes or type 2 diabetes for their AhR transactivation activity (AhRTA). To our surprise, the AhRT activities of sera were closely associated with the parameters of the metabolic syndrome. Results were consistent with the conclusion that serum dioxin-like activity is a "cause" of insulin resistance and type 2 diabetes. In short, AhRT activities and parameters of the metabolic syndrome were linear (Submitted for publication). This linear relationship is in stark contrast with the conclusion obtained from CARDIA cohorts, where a "low dose effect" predominates. I wonder if this study could be extended to a nested case control study like CARDIA.
BIOASSAY OF SERUM DIOXIN-LIKE ACTIVITY, A BREAKTHROUGH?
- Critics of the mitochondria-based model argue that excessive calorie intake and lack of exercise are the primary causes of obesity, and thus the metabolic syndrome. It is frequently argued that even though mitochondrial dysfunction is present in this state, it is regarded as secondary to obesity, induced by overfeeding or lack of exercise. Indeed, one can surely bring on obesity by consuming a high-fat diet. However, these critiques dismiss some important facts; 1) only certain high-fat diets cause obesity in susceptible animals, (this "high-fat diet" was found to be contaminated with POPs [unpublished observation]); 2) the propensity to return to previous body weight after fat removal or transplantation suggests the presence of a strong drive to maintain body weight; and 3) obese people do not eat more, once a certain increase in body weight is reached. I will not provide references supporting all of these facts, but I want to emphasize that the current gold standard of diet assessment, the doubly labeled water test, which is held to provide unequivocal evidence for excessive calorie intake as a cause of obesity, might be based on an erroneous assumption.
- A basic assumption of the doubly labeled water (DLW) method to measure energy expenditure is that H of body water only leaves the body pool in the form of water. If deuterium (as a measure of H) is sequestered in anything other than body water during a DLW study, an overestimation of water turnover and therefore an underestimation of CO2 production would result, thus indicating a high metabolic rate. Hydrogen is incorporated into fat, and deuterium incorporation into body fat has the greatest potential to be an error of the DLW method. This is significant and presents a potentially serious error in the determination of energy requirements, especially in those subjects with different intrinsic rates of fat synthesis. Haggarty et al. [17] reported de novo lipogenesis and cholesterol synthesis over the typical duration of a DLW study (2 weeks), as well as the likely effects of sequestration on the performance of the DLW method in adult human subjects.
- Using whole-body fatty acid synthesis, they found little evidence of an error on DLW-derived CO2 production for five of the six subjects studied; mean -0×5%, range 0 to -1×3%. However, there was a subject whose fatty acid synthesis rate was almost 10 times faster than others, was hypertrigliceridemic (five times higher) and had a slightly higher body fat mass. They argued that the very high value for circulating triacylglycerols in this subject was indicative of a high rate of de novo lipid synthesis and that the assumptions underlying the calculation of whole-body lipogenesis (assumed identical between subjects) may be invalid. In other words, in subjects with hypertriglyceridemia, the DLW test might result in rather high estimates of energy expenditure.
- In the metabolic syndrome (with hyertriglyceridemia), a DLW test would result in a high estimate of energy expenditure compared with a diet intake measure (which is normal for the body mass). One would arrive at an erroneous conclusion that obese subjects under-report their dietary intake.
CRITIQUES OF THE MITOCHONDRIAL HYPOTHESIS
- I presented evidence that POPs might be a cause of the metabolic syndrome, probably acting through AhR and a mitochondrial mechanism. There is a huge body of evidence supporting this hypothesis. However, establishing a cause and effect relation between POP exposure and the development of the metabolic syndrome will be difficult, because of the diversity of POPs and their bizarre low dose effect. This conundrum might be resolved by our finding that serum AhRT activity is linearly related with parameters of the metabolic syndrome in nested case control studies.
- The mitochondria-based model could be considered as a special application of the metabolic scaling law as applied to a group of people with a specific disease. This model will be very useful in integrating various factors acting on mitochondrial function, such as genes, fetal malnutrition and environmental toxins and protectors. I believe that "genomic medicine" should be based on the mitochondrial genome, not only the nuclear genome, as our model suggests. Our model also suggests that if we could improve mitochondrial function, insulin resistance might decrease. This prediction is validated by recent studies with hydrogen gas, which quenches free radicals and stimulates mitochondrial function [18], and by studies with bile acid sequestrants, which could remove POPs and improve the metabolic syndrome [19].
SUMMARY AND CONCLUSIONS
-
Acknowledgements
- In receiving this prestigious award from the Korean Diabetes Association, I want to thank my mentors, professors Eung-Jin Kim, Munho Lee, and Hun-Ki Min, at Seoul National University College of Medicine, for their teaching and support of my personal and scientific development. I also want to thank my colleagues and students at Seoul National University College of Medicine, Korean NIH, and Eulji University and Hospital, for various contributions to the development of the mitochondrial hypothesis. I could not name all but a few; Chan-Soo Shin, Yong-Soo Park, Ki-Up Lee, Kyong-Soo Park, Hak-Chul Jang, Young-Min Cho, Soo Lim, Ji-Hyun Song, Young-Mi Kim, Eun-Bo Shim, Nam-Han Cho, Jin-Taek Kim, and Dae-Won Jun. I also want to thank Professor Kishio Najo at Wakayama University, Dr. Masashi Tanaka at Tokyo Metropolitan Institute of Ageing, both of Japan, Professor Yau-Huei Wei, at McKay University, Taiwan, Professors Jang-Hyun Yoon at the University of Southern California, Ronald E LaPorte at the University of Pittsburgh, and Thomas T Aoki at the University California at Davis, of USA, for their helpful discussions and support over the years.
ACKNOWLEDGMENTS
- 1. Lim S, Cho YM, Park KS, Lee HK. Persistent organic pollutants, mitochondrial dysfunction, and metabolic syndrome. Ann N Y Acad Sci 2010;1201:166-176. ArticlePubMed
- 2. Lee HK, Cho YM, Kwak SH, Lim S, Park KS, Shim EB. Mitochondrial dysfunction and metabolic syndrome-looking for environmental factors. Biochim Biophys Acta 2010;1800:282-289. ArticlePubMed
- 3. Savage VM, Allen AP, Brown JH, Gillooly JF, Herman AB, Woodruff WH, West GB. Scaling of number, size, and metabolic rate of cells with body size in mammals. Proc Natl Acad Sci U S A 2007;104:4718-4723. ArticlePubMedPMC
- 4. Weibel ER. Physiology: the pitfalls of power laws. Nature 2002;417:131-132. ArticlePubMedPDF
- 5. Fuku N, Park KS, Yamada Y, Nishigaki Y, Cho YM, Matsuo H, Segawa T, Watanabe S, Kato K, Yokoi K, Nozawa Y, Lee HK, Tanaka M. Mitochondrial haplogroup N9a confers resistance against type 2 diabetes in Asians. Am J Hum Genet 2007;80:407-415. ArticlePubMedPMC
- 6. Cho YM, Park KS, Lee HK. Genetic factors related to mitochondrial function and risk of diabetes mellitus. Diabetes Res Clin Pract 2007;77(Suppl 1):S172-S177. ArticlePubMed
- 7. Palmieri VO, De Rasmo D, Signorile A, Sardanelli AM, Grattagliano I, Minerva F, Cardinale G, Portincasa P, Papa S, Palasciano G. T16189C mitochondrial DNA variant is associated with metabolic syndrome in Caucasian subjects. Nutrition 2011;27:773-777. ArticlePubMed
- 8. Wilson FH, Hariri A, Farhi A, Zhao H, Petersen KF, Toka HR, Nelson-Williams C, Raja KM, Kashgarian M, Shulman GI, Scheinman SJ, Lifton RP. A cluster of metabolic defects caused by mutation in a mitochondrial tRNA. Science 2004;306:1190-1194. ArticlePubMedPMC
- 9. Lee YY, Park KS, Pak YK, Lee HK. The role of mitochondrial DNA in the development of type 2 diabetes caused by fetal malnutrition. J Nutr Biochem 2005;16:195-204. ArticlePubMed
- 10. Baillie-Hamilton PF. Chemical toxins: a hypothesis to explain the global obesity epidemic. J Altern Complement Med 2002;8:185-192. ArticlePubMed
- 11. Lee DH, Lee IK, Song K, Steffes M, Toscano W, Baker BA, Jacobs DR Jr. A strong dose-response relation between serum concentrations of persistent organic pollutants and diabetes: results from the National Health and Examination Survey 1999-2002. Diabetes Care 2006;29:1638-1644. PubMed
- 12. Lee DH, Lee IK, Porta M, Steffes M, Jacobs DR Jr. Relationship between serum concentrations of persistent organic pollutants and the prevalence of metabolic syndrome among non-diabetic adults: results from the National Health and Nutrition Examination Survey 1999-2002. Diabetologia 2007;50:1841-1851. ArticlePubMedPDF
- 13. Lee DH, Steffes MW, Sjodin A, Jones RS, Needham LL, Jacobs DR Jr. Low dose of some persistent organic pollutants predicts type 2 diabetes: a nested case-control study. Environ Health Perspect 2010;118:1235-1242. ArticlePubMedPMC
- 14. Lim S, Ahn SY, Song IC, Chung MH, Jang HC, Park KS, Lee KU, Pak YK, Lee HK. Chronic exposure to the herbicide, atrazine, causes mitochondrial dysfunction and insulin resistance. PLoS One 2009;4:e5186ArticlePubMedPMC
- 15. Ruzzin J, Petersen R, Meugnier E, Madsen L, Lock EJ, Lillefosse H, Ma T, Pesenti S, Sonne SB, Marstrand TT, Malde MK, Du ZY, Chavey C, Fajas L, Lundebye AK, Brand CL, Vidal H, Kristiansen K, Froyland L. Persistent organic pollutant exposure leads to insulin resistance syndrome. Environ Health Perspect 2010;118:465-471. ArticlePubMed
- 16. Lee DH, Steffes MW, Sjodin A, Jones RS, Needham LL, Jacobs DR Jr. Low dose organochlorine pesticides and polychlorinated biphenyls predict obesity, dyslipidemia, and insulin resistance among people free of diabetes. PLoS One 2011;6:e15977ArticlePubMedPMC
- 17. Haggarty P, Shetty P, Thangam S, Kumar S, Kurpad A, Ashton J, Milne E, Earl C. Free and esterified fatty acid and cholesterol synthesis in adult males and its effect on the doubly-labelled water method. Br J Nutr 2000;83:227-234. ArticlePubMed
- 18. Kamimura N, Nishimaki K, Ohsawa I, Ohta S. Molecular hydrogen improves obesity and diabetes by inducing hepatic FGF21 and stimulating energy metabolism in db/db mice. Obesity (Silver Spring) Epub 2011 Feb 3. DOI:10.1038/oby.2011.6.ArticlePDF
- 19. Yamaoka-Tojo M, Tojo T, Izumi T. Beyond cholesterol lowering: pleiotropic effects of bile acid binding resins against cardiovascular disease risk factors in patients with metabolic syndrome. Curr Vasc Pharmacol 2008;6:271-281. ArticlePubMed
REFERENCES
Fig. 1Pathogenesis of the metabolic syndrome as a special state of mitochondrial dysfunction. Various phenotypes, such as insulin deficiency and insulin resistance are pathophysiologically defined states, while type 2 diabetes, hypertension and obesity are well-defined specific disease states. Coronary heart disease and cancer might be considered either specific diseases or complications (or more appropriately, later stages) of preceding disease states. In this model, diabetes and CADs are not results of obesity, but its later stage phenotypes. CVD, cardiovascular disease; NMD, neuromuscular disease; NAFLD, nonalcoholic fatty liver disease.
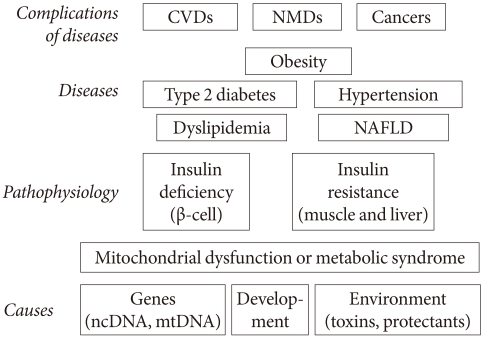
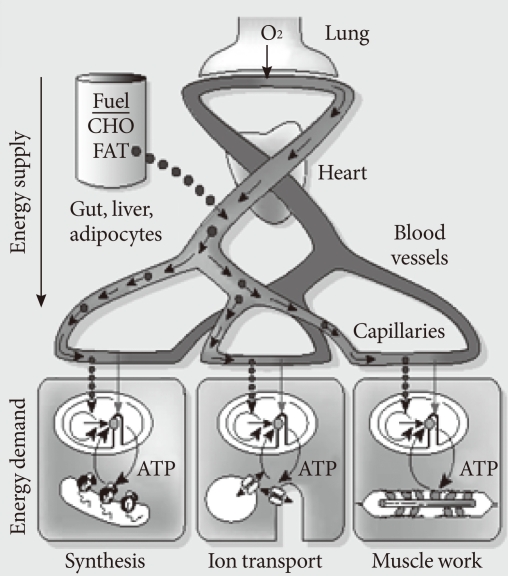
Fig. 2Metabolic scaling relation between body size and metabolic rate. Panel (A) shows metabolic rate increases (watt) with increasing body mass (kg) across animal species, from mitochondrion to elephant. In Panel (B), the logarithm of the mass-specific metabolic rate, W/g, versus the body mass, M (g), for mammals is plotted in a logarithmic scale. The mass-specific metabolic rate declines as body mass increases. Panel (C) shows the relationship between changes in 24-hour energy expenditure (Δ 24-EE) and Δ weight after adjustment of 24-EE for fat-free mass (FFM), fat mass (FM), waist to thigh ratio (WTR), and age. Weight change is accompanied by a change in energy expenditure in Pima Indians. A long-term adaptation process normalizes the relationship between body mass and metabolic rate, suggesting that an adaptive process is operating (Panel D). Arrows 1A and 2A depicts metabolic adaptation, which leads to a quick decrease in the metabolic drive and a small weight change. Arrows 1B and 2B depict poor metabolic adaptation, which leads to a sustained metabolic drive and a larger weight change.

Fig. 3In whole-body metabolism, energy supply and energy demand are tightly linked, and the metabolism of its parts is quantitatively related (allometry) to whole-body mass. The larger the body, the bigger the parts, but the mass specific metabolic rate decreases (From Weibel ER. Nature 2002;417:131-2, with permission from Nature Publishing Group) [4].
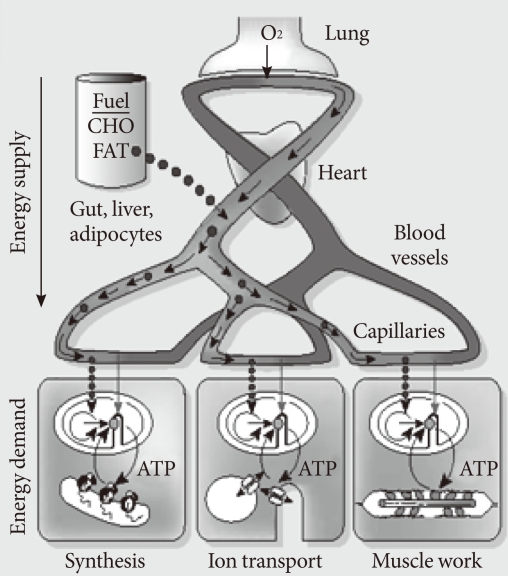
Figure & Data
References
Citations
Citations to this article as recorded by 
- Drug-induced mitochondrial toxicity: Risks of developing glucose handling impairments
Auxiliare Kuretu, Charles Arineitwe, Mamosheledi Mothibe, Phikelelani Ngubane, Andile Khathi, Ntethelelo Sibiya
Frontiers in Endocrinology.2023;[Epub] CrossRef - Obesogens in Foods
Iva Kladnicka, Monika Bludovska, Iveta Plavinova, Ludek Muller, Dana Mullerova
Biomolecules.2022; 12(5): 680. CrossRef - Pesticides and pancreatic adenocarcinoma: A transversal epidemiological, environmental and mechanistic narrative review
Mathias Brugel, Claire Carlier, Gabriela Reyes-Castellanos, Sidonie Callon, Alice Carrier, Olivier Bouché
Digestive and Liver Disease.2022; 54(12): 1605. CrossRef - Low-Dose Dioxin Reduced Glucose Uptake in C2C12 Myocytes: The Role of Mitochondrial Oxidative Stress and Insulin-Dependent Calcium Mobilization
Suyeol Im, Sora Kang, Ji Hwan Kim, Seung Jun Oh, Youngmi Kim Pak
Antioxidants.2022; 11(11): 2109. CrossRef - Hallmarks of Health
Carlos López-Otín, Guido Kroemer
Cell.2021; 184(1): 33. CrossRef - Prenatal exposure to persistent organic pollutants and markers of obesity and cardiometabolic risk in Spanish adolescents
Nuria Güil-Oumrait, Damaskini Valvi, Raquel Garcia-Esteban, Monica Guxens, Jordi Sunyer, Maties Torrent, Maribel Casas, Martine Vrijheid
Environment International.2021; 151: 106469. CrossRef - Clinical Value of Serum Mitochondria-Inhibiting Substances in Assessing Renal Hazards: A Community-Based Prospective Study in Korea
Hoon Sung Choi, Jin Taek Kim, Hong Kyu Lee, Wook Ha Park, Youngmi Kim Pak, Sung Woo Lee
Endocrinology and Metabolism.2021; 36(6): 1298. CrossRef - Potential role of aryl hydrocarbon receptor signaling in childhood obesity
Nancy N. Shahin, Ghada T. Abd-Elwahab, Afaf A. Tawfiq, Hanan M. Abdelgawad
Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids.2020; 1865(8): 158714. CrossRef - Potential contribution of insecticide exposure and development of obesity and type 2 diabetes
Xiao Xiao, John M. Clark, Yeonhwa Park
Food and Chemical Toxicology.2017; 105: 456. CrossRef - Persistent Organic Pollutants as Risk Factors for Obesity and Diabetes
Chunxue Yang, Alice Pik Shan Kong, Zongwei Cai, Arthur C.K. Chung
Current Diabetes Reports.2017;[Epub] CrossRef - Aryl hydrocarbon receptor deficiency protects mice from diet-induced adiposity and metabolic disorders through increased energy expenditure
C-X Xu, C Wang, Z-M Zhang, C D Jaeger, S L Krager, K M Bottum, J Liu, D-F Liao, S A Tischkau
International Journal of Obesity.2015; 39(8): 1300. CrossRef - Disruption of Ah Receptor Signaling during Mouse Development Leads to Abnormal Cardiac Structure and Function in the Adult
Vinicius S. Carreira, Yunxia Fan, Hisaka Kurita, Qin Wang, Chia-I Ko, Mindi Naticchioni, Min Jiang, Sheryl Koch, Xiang Zhang, Jacek Biesiada, Mario Medvedovic, Ying Xia, Jack Rubinstein, Alvaro Puga, Diego Fraidenraich
PLOS ONE.2015; 10(11): e0142440. CrossRef - Persistent organic pollutants and biomarkers of diabetes risk in a cohort of Great Lakes sport caught fish consumers
Mary Turyk, Giamila Fantuzzi, Victoria Persky, Sally Freels, Anissa Lambertino, Maria Pini, Davina H. Rhodes, Henry A. Anderson
Environmental Research.2015; 140: 335. CrossRef - Serum aryl hydrocarbon receptor ligand activity is associated with insulin resistance and resulting type 2 diabetes
Eun Roh, Soo Heon Kwak, Hye Seung Jung, Young Min Cho, Youngmi Kim Pak, Kyong Soo Park, Seong Yeon Kim, Hong Kyu Lee
Acta Diabetologica.2015; 52(3): 489. CrossRef - A cross-sectional analysis of dioxins and health effects in municipal and private waste incinerator workers in Japan
Kenya YAMAMOTO, Mitsuhiro KUDO, Heihachiro ARITO, Yasutaka OGAWA, Tsutomu TAKATA
INDUSTRIAL HEALTH.2015; 53(5): 465. CrossRef - Association of Dioxin and Other Persistent Organic Pollutants (POPs) with Diabetes: Epidemiological Evidence and New Mechanisms of Beta Cell Dysfunction
Vincenzo De Tata
International Journal of Molecular Sciences.2014; 15(5): 7787. CrossRef - The Glycated Albumin to Glycated Hemoglobin Ratio Might Not Be Associated with Carotid Atherosclerosis in Patients with Type 1 Diabetes
Wonjin Kim, Kwang Joon Kim, Byung-Wan Lee, Eun Seok Kang, Bong Soo Cha, Hyun Chul Lee
Diabetes & Metabolism Journal.2014; 38(6): 456. CrossRef - Pesticides and human chronic diseases: Evidences, mechanisms, and perspectives
Sara Mostafalou, Mohammad Abdollahi
Toxicology and Applied Pharmacology.2013; 268(2): 157. CrossRef - Coenzyme Q10 Ameliorates Pain and Cartilage Degradation in a Rat Model of Osteoarthritis by Regulating Nitric Oxide and Inflammatory Cytokines
Jennifer Lee, Yeon Sik Hong, Jeong Hee Jeong, Eun Ji Yang, Joo Yeon Jhun, Mi Kyoung Park, Young Ok Jung, Jun Ki Min, Ho Youn Kim, Sung Hwan Park, Mi-La Cho, Zoltan Rakonczay
PLoS ONE.2013; 8(7): e69362. CrossRef - Plasmatic concentration of organochlorine lindane acts as metabolic disruptors in HepG2 liver cell line by inducing mitochondrial disorder
Mohammed el Amine Benarbia, David Macherel, Sébastien Faure, Caroline Jacques, Ramaroson Andriantsitohaina, Yves Malthièry
Toxicology and Applied Pharmacology.2013; 272(2): 325. CrossRef - Increased Susceptibility to Metabolic Syndrome in Adult Offspring of Angiotensin Type 1 Receptor Autoantibody-Positive Rats
Suli Zhang, Xi Zhang, Lihong Yang, Zi Yan, Li Yan, Jue Tian, Xiaoyu Li, Li Song, Li Wang, Xiaoli Yang, Ronghua Zheng, Wayne Bond Lau, Xinliang Ma, Huirong Liu
Antioxidants & Redox Signaling.2012; 17(5): 733. CrossRef
 PubReader
PubReader Cite
Cite