- Current
- Browse
- Collections
-
For contributors
- For Authors
- Instructions to authors
- Article processing charge
- e-submission
- For Reviewers
- Instructions for reviewers
- How to become a reviewer
- Best reviewers
- For Readers
- Readership
- Subscription
- Permission guidelines
- About
- Editorial policy
Articles
- Page Path
- HOME > Diabetes Metab J > Volume 36(3); 2012 > Article
-
ReviewComplications Reactive Oxygen and Nitrogen Species in Pathogenesis of Vascular Complications of Diabetes
- Seok Man Son
-
Diabetes & Metabolism Journal 2012;36(3):190-198.
DOI: https://doi.org/10.4093/dmj.2012.36.3.190
Published online: June 14, 2012
Department of Internal Medicine, Pusan National University School of Medicine, Yangsan, Korea.
- Corresponding author: Seok Man Son. Department of Internal Medicine, Pusan National University School of Medicine and Diabetes Center and Endocrine Clinic, Pusan National University Yangsan Hospital, 20 Geumo-ro, Mulgeum-eup, Yangsan 626-770, Korea. sonsm@pusan.ac.kr
Copyright © 2012 Korean Diabetes Association
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- Macrovascular and microvascular diseases are currently the principal causes of morbidity and mortality in subjects with diabetes. Disorders of the physiological signaling functions of reactive oxygen species (superoxide and hydrogen peroxide) and reactive nitrogen species (nitric oxide and peroxynitrite) are important features of diabetes. In the absence of an appropriate compensation by the endogenous antioxidant defense network, increased oxidative stress leads to the activation of stress-sensitive intracellular signaling pathways and the formation of gene products that cause cellular damage and contribute to the vascular complications of diabetes. It has recently been suggested that diabetic subjects with vascular complications may have a defective cellular antioxidant response against the oxidative stress generated by hyperglycemia. This raises the concept that antioxidant therapy may be of great benefit to these subjects. Although our understanding of how hyperglycemia-induced oxidative stress ultimately leads to tissue damage has advanced considerably in recent years, effective therapeutic strategies to prevent or delay the development of this damage remain limited. Thus, further investigation of therapeutic interventions to prevent or delay the progression of diabetic vascular complications is needed.
- Diabetes diagnoses are increasing at an alarming rate worldwide. The majority of diabetes-related deaths arise from vascular complications such as myocardial infarction, cerebrovascular disease, and peripheral vascular disease [1]. Large prospective randomized clinical studies show that long-term glycemic control is an important predictor of diabetic vascular complications [1,2].
- There is considerable evidence that many of the biochemical pathways adversely affected by hyperglycemia including glucose oxidation, the formation of advanced glycation end-products (AGE), and activation of polyol pathways, are associated with the generation of reactive oxygen species (ROS), ultimately leading to increased oxidative stress in a variety of tissues [3]. In the absence of an appropriate compensation by the endogenous antioxidant defense network, increased oxidative stress leads to the activation of stress-sensitive intracellular signaling pathways and the formation of gene products that cause cellular damage and contribute to the late complications of diabetes [3,4].
- Major reactive species, including ROS and reactive nitrogen species (RNS) consist of paramagnetic free radicals (superoxide O2•-, hydroxyl HO•, peroxy ROO•, and nitric oxide free radical NO•) and diamagnetic molecules (hydrogen peroxide H2O2 and peroxynitrite ONOO-) which are products of the reactions of these free radicals. All these species were previously considered to be toxic agents capable of damaging biomolecules. However it is now known that physiological free radicals superoxide and nitric oxide are relatively harmless species that are able to initiate or mediate many enzyme- and gene-dependent reactions in both physiological and pathophysiological processes. Increasing evidence in experimental and clinical studies suggests that ROS/RNS play important roles in the pathogenesis of diabetic vascular diseases [4].
- This review summarizes recent knowledge on the relevance of oxidative and nitrosative stress in the pathophysiology of diabetic vascular complications.
INTRODUCTION
- Sources of ROS
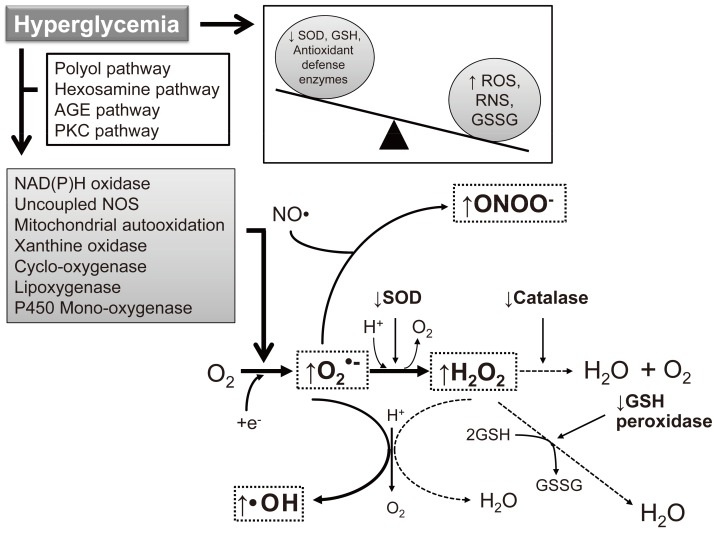
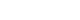
- As the development of diabetes mellitus is characterized by high serum glucose levels, this prooxidant molecule can be an origin of ROS overproduction (Fig. 1). High glucose (HG) levels can initiate the production of superoxide and hydrogen peroxide, precursors of reactive free radicals, which are able to stimulate the decline of antioxidant systems, directly damage many biomolecules, and increase lipid peroxidation in diabetes [5,6].
- The most important sources of ROS under hyperglycemic conditions are mitochondria and NAD(P)H oxidases. Hyperglycemia decreases glyceraldehyde-3-phosphate dehydrogenase (GAPDH) activity in bovine aortic endothelial cells via the increased production of mitochondrial superoxide and a concomitant increase in hexosamine pathway activity [7,8]. Mitochondrial ROS have also been implicated in diabetic complications and progression of the underlying diabetic state; however, it is not clear whether mitochondria of diabetic origin actually generate ROS independent of the surrounding diabetic milieu.
- NAD(P)H oxidases are another important ROS source in diabetes. Superoxide production significantly increases by upregulated NAD(P)H oxidase and endothelial NO synthase in human blood vessels from subjects with type 2 diabetes [9]. These effects of NAD(P)H oxidase and uncoupling eNOS might be mediated by protein kinase C (PKC) signaling. Our group assumed that the enhanced proliferative capacity of diabetic vascular smooth muscle cells (VSMC) is a consequence of NAD(P)H oxidase activation stimulated by the activation of a protein tyrosine kinase [10]. It has been suggested that ROS formation by HG-stimulated NAD(P)H oxidase can also upregulate antioxidant enzymes [11]. Therefore NAD(P)H oxidase might be a double-edged sword, providing feedback defense against excessive ROS generation through the activation of receptor tyrosine kinases and the redox-sensitive transcription factor Nrf2-Keap1 signaling pathway [11]. It is of importance that Block et al. [12] have recently identified NAD(P)H oxidase (Nox4) as mitochondrial cytochrome c oxidase subunit IV as being an additional source of ROS in diabetes. It should be noted that xanthine oxidase is also a potential source of ROS formation in diabetes.
- ROS overproduction in diabetes
- Diabetes causes increased oxidative stress in various tissues as evidenced by increased levels of oxidized DNA, proteins, and lipids. Besides damaging the functions of these molecules, oxidative stress also triggers a series of cellular responses, including the activation of PKC, NF-κB, and JNK stress-associated kinases [13]. Inappropriate activation of these important regulatory molecules can have deleterious effects on cellular functions, and is thought to contribute to the pathogenesis of various diabetic vascular complications. However, it is not clear how hyperglycemia leads to increased oxidative stress. Most likely, both the increased production of ROS and decreased capacity of the cellular antioxidant defense system combine to produce oxidative stress in diabetes.
- Hyperglycemia increases oxidant production by multiple pathways rather than a single dominant pathway. Glucose can undergo non-enzymatic reactions to form gluco-oxidants and glycated products, which can be oxidants [13]. Metabolism of excessive intracellular glucose can occur by several processes such as aldose reductase, mitochondrial oxidative phosphorylation, activation of NAD(P)H oxidases, and alteration of NADPH/NADP ratios [3]. Among these possibilities, recent work has centered on mitochondrial metabolism and activation of NAD(P)H oxidases [3,13]. It has been suggested that most glucose-induced oxidants are derived from overproduction of superoxide by the mitochondrial electron-transport chain [3]. By-products of this process will trigger signaling cascades such as activation of PKC, production of hexosamine, increased flux via aldose reductase, and production of glycated products. However, other investigators have reported that high glucose levels can activate NAD(P)H oxidases in vascular cells independent of mitochondrial metabolism [14]. It is known that the ROS activation of PKC stimulates subsequent ROS overproduction; therefore this enzymatic cascade can be an important harmful signaling process in hyperglycemia and diabetes. Thus, Koya and King [15] suggested that glucose's adverse effects in diabetes and hyperglycemia might be a consequence of ROS-stimulated activation of the diacylglycerol (DAG)/PKC enzymatic pathway because treatment with α-tocopherol prevented glucose-induced vascular dysfunctions and inhibited DAG/PKC activation. ROS signaling is also an important stimulus of harmful enzymatic cascades catalyzed by protein kinase Akt/B [16].
- Recently, two other aspects have been found to be related to glucose-induced oxidative stress. First, several studies have suggested that intermittent low and high glucose conditions (a condition known as "glucose variability") are even more deleterious to endothelial cell function than a steady, constant increase in glucose [17]. These conditions also induce endothelial cells to enter into a proinflammatory state, and this state is associated with the upregulation of various adhesion molecules and proinflammatory cytokines [17]. Some of the pathways implicated in these exacerbated cellular responses, involve activation of PKC, NAD(P)H oxidases, and mitochondrial oxidants. Secondly, it has been shown in culture and in diabetic rats that endothelial cells exhibit a persistence or 'memory' of induced basement membrane mRNA expression long after normalization of high glucose levels, a condition called "glucose memory" [18]. This finding suggests that glucose induces long-lasting deleterious effects that persist beyond the period of hyperglycemia.
- Abundant evidence exists that many protein, lipid, and DNA markers of oxidative stress are increased in cultured vascular cells exposed to high glucose levels and in vascular tissues from animals and human subjects with diabetes [13]. In cultured vascular cells, elevating glucose levels in the media increases the production of oxidants such as gluco-oxidants, glycated compounds, oxidized low density lipoprotein (LDL), superoxide, and nitrotyrosine [14]. Similarly, elevated levels of isoprostanes, 8-hydroxydeoxyguanosine, and lipid peroxides have been reported in diabetic animals and in human subjects with diabetes [14]. Although numerous reports have substantiated that oxidant production is increased in diabetes, clinical evidence for tissue damage as a result of oxidative stress has not been clearly demonstrated because both plasma and cells contain a large reserve of antioxidants. In fact, the levels of various antioxidants in plasma and cells have not consistently been shown to decrease in diabetic states [14].
- Role of ROS in development of macrovascular complications
- Abnormalities in endothelial and vascular smooth muscle cell function, as well as the coagulation system, contribute to vascular complications in diabetes.
- Vascular endothelial dysfunction
- Endothelial cells modulate vascular function and structure. In normal endothelial cells, nitric oxide is synthesized and released to maintain vascular homeostasis. Clinical studies have consistently found that endothelium-dependent vasodilation is abnormal in subjects with diabetes [15]. The major contributor to endothelial oxidative stress is the increased production of superoxide. This seems to occur via two principal sources: NAD(P)H oxidases and uncoupled endothelial nitric oxide synthase (eNOS). Hyperglycemia, AGE, free fatty acid (FFA), and oxidized LDL have been shown to increase endothelial NAD(P)H oxidase activity and the activation of NAD(P)H oxidases by hyperglycemia and FFA has been shown to be mediated by PKC [16]. Vessels isolated from diabetic subjects exhibit increased superoxide production that is inhibited by diphenylene iodinium, and demonstrate increased expression of several NAD(P)H oxidase subunits (p22phox, p47phox, and p67phox) [17], suggesting that NAD(P)H oxidases are more active in diabetes. Not only does excess superoxide itself cause increased oxidative stress, but it can also react with nitric oxide (NO•) to produce peroxynitrite [18], which, in turn, can oxidize tetrahydrobiopterin (BH4), thus reducing its availability to eNOS [17,18]. In the presence of reduced concentrations of BH4, eNOS becomes uncoupled and transfers electrons to molecular oxygen instead of L-arginine to produce superoxide rather than NO•. The presence of uncoupled eNOS in the diabetic vasculature is supported by a study in which diabetic vessels were found to produce less superoxide when incubated with the NO• synthase inhibitor NG-nitro-L-arginine methyl ester [17]. BH4 availability may also be decreased by a reduction of its synthesis. The expression of GTP-cyclohydrolase I (GTPCH), the rate-limiting enzyme for de novo BH4 synthesis, is reduced in diabetic rats [19]. Furthermore, transgenic mice overexpressing GTPCH treated with streptozotocin (STZ) are able to maintain endothelial function [19]. Clinical studies have demonstrated that BH4 supplementation given to diabetic subjects improves their endothelium-dependent vasodilation, indicating that uncoupled eNOS plays a role in diabetic endothelial dysfunction [17-19].
- An additional effect of high serum glucose levels is delayed replication of large-vessel endothelium. Superoxide dismutase, catalase, and reduced glutathione protect human endothelial cells from glucose-induced delay in replication, offering further evidence of the importance of oxidative stress in diabetes [20].
- Vascular smooth muscle dysfunction
- In type 2 diabetic subjects, the vasodilator response to endogenous nitric oxide donors is diminished [15,20], suggesting that there is a fundamental abnormality in VSMC function. The oxidative stress produced in VSMC in diabetes may shift them from a contractile to a proliferative phenotype, thus further inhibiting vasodilation and enhancing lesion formation. Subjects with diabetes have increased proliferation and migration of VSMC into atherosclerotic lesions [21]. There are a number of studies showing that exposure of VSMC to high glucose conditions results in oxidative stress and subsequent cell proliferation.
- Hyperglycemia causes PKC activation and subsequent ROS production via NAD(P)H oxidase in cultured aortic smooth muscle cells [14]. In STZ-treated rats, a p22phox-containing NAD(P)H oxidase was found to be a mediator of VSMC proliferation [22]. Additionally, the polyol pathway has been implicated in hyperglycemia-induced, PKC-directed NF-κB activation, as inhibition of aldose reductase is able to mitigate both PKC and NF-κB activation in cultured rat aortic smooth muscle cells [23]. Despite the increased proliferation and migration of VSMC in subjects with diabetes [21], atherosclerotic lesions from these patients have fewer VSMC, suggesting that VSMC death potentially plays a role in plaque instability and subsequent rupture [24]. A study in human aortic smooth muscle cells revealed that high glucose conditions cause cell necrosis via hydrogen peroxide [24]. Additionally, oxidized LDL is able to induce VSMC apoptosis via ROS and may be increased in diabetic subjects [24].
- In diabetic subjects, the elaboration of cytokines diminishes synthesis collagen of vascular smooth muscle and increases production of matrix metalloproteinases, which may also lead to an increased tendency for plaque destabilization and rupture [21,24].
- Role of ROS in development of microvascular complications
- The microvascular complications of diabetes, including nephropathy, retinopathy and neuropathy, are common manifestations of diabetes. Although the mechanisms underlying the development of these conditions are incompletely understood, oxidative stress has been implicated. In isolated kidneys, exposure to ROS causes a drastic dose-dependent decrease in de novo synthesis of heparan sulfate, an effect that is reversed by catalase [25]. In addition, the ability of glomeruli isolated from STZ-induced diabetic rats to degrade hydrogen peroxide is greatly impaired; this has been attributed to either decreased catalase activity or altered glutathione redox cycling [25]. Recent data has shown that the expression of the NAD(P)H oxidase subunits Nox4 and p22phox are upregulated in the kidney of STZ-induced diabetic rats and that NAD(P)H oxidase-dependent production of ROS may cause DNA damage in diabetic renal tissues leading to the development of nephropathy [26].
- Evidence for the participation of oxidative stress in the pathogenesis of diabetic retinopathy is scant and limited to studies published in abstract form. One report showed that the activity of NAD(P)H oxidase was increased in the retina of diabetic rats and that this might be involved in the development of diabetic retinopathy [27].
- Stronger evidence exists for an involvement of ROS in the etiology of early experimental diabetic neuropathy. One report showed that probucol treatment prevents the reduction in nerve conduction velocity in STZ-induced diabetic rats and in normal rats treated with primaquine. The authors suggested that nerve dysfunction depends on oxidative stress that causes neurovascular defects resulting in endoneural hypoxia [28]. Treatment with the transition-metal chelating agents deferoxamine and trientine, which can prevent auto-oxidation, corrected nerve conduction and blood flow changes that occurred 2 months after the induction of diabetes in this model [28].
REACTIVE OXYGEN SPECIES SIGNALING IN DIABETES
- Nitric oxide is an endothelium-derived relaxing factor (EDRF) and its role is very important in the normal activity of cells. Therefore, it might be expected that as in other pathological states a decrease in nitric oxide can take place in diabetes. On the other hand, nitric oxide is soluble in aqueous and lipid media and it rapidly diffuses through the cytoplasm and plasma membranes. In an inflammatory state, such as obesity, the immune system produces both superoxide and nitric oxide, which may react together to produce significant amounts of peroxynitrite anion (ONOO-). Peroxynitrite is a potent oxidizing agent that can cause DNA fragmentation and lipid peroxidation [29]. There are multiple lines of evidence demonstrating the formation of peroxynitrite in diabetic vasculature, both in experimental models and in humans [30,31]. The tissues and species in which peroxynitrite has been identified in experimental animals and in humans include plasma, kidney, blood vessels, retina, heart and peripheral nerves and have been overviewed recently [30]. The mechanisms that underlie peroxynitrite-induced diabetic complications and vascular alterations are multiple [31,32]. One of the important pathways of peroxynitrite-mediated vascular dysfunction in diabetes involves activation of the nuclear enzyme poly(ADP-ribose) polymerases (PARP enzymes). Activated PARP-1 cleaves NAD+ into nicotinamide and ADPribose and polymerizes the latter on nuclear acceptor proteins. Peroxynitrite-induced overactivation of PARP consumes NAD+ and consequently ATP culminating in cell dysfunction, apoptosis or necrosis [33]. Thus Langenstroer and Pieper [34] found that spontaneous EDRF released in diabetic rat aorta can be unmasked by the addition of superoxide dismutase (SOD) because SOD produces a significantly greater relaxation in diabetic aorta compared to control aorta. It means that NO synthases in diabetes produce superoxide in an uncoupled state. Other authors have also suggested that peroxynitrite formed by uncoupled NO synthases in human blood vessels from subjects with diabetes type 2 can be a mediator of the cytotoxic effects of high glucose [35].
RNS SIGNALING IN DIABETES
- Many trials in diabetic subjects and animal models of diabetes have attempted to determine whether antioxidant treatments can prevent or delay the onset of diabetic complications.
- If the effects of ROS/RNS signaling in enzyme/gene processes depend on the levels of ROS/RNS production, then ROS/RNS-induced damage in diabetes as well in the other pathologies such as cardiovascular diseases, inflammation, and aging cannot be successfully inhibited by the traditional antioxidants vitamin C or vitamin E which are unable to react with superoxide [36,37]. Nonetheless some aforementioned data demonstrate the successful application of various antioxidants and free radical scavengers for suppression of damaging processes in diabetes.
- Experimental animal trials
- Several antioxidants have been studied in diabetic and insulin-resistant animals, mostly rodents, with respect to their ability to correct vascular and neurologic dysfunction [28].
- α-Lipoic acid is needed to regenerate glutathione and oxidized vitamins C and E. In animal studies, vitamins C and E and α-lipoic acid improve nerve conduction velocity and blood flow to the peripheral nerves, reduce retinal leukocyte adhesions, and improve cataract formation, suggesting that they may be an effective treatment of diabetic complications [28,38]. Furthermore, vitamins C and E, either individually or in combination, normalize many parameters of oxidative stress such as lipid peroxidation, isoprostane production, plasma malondialdehyde, and NF-κB activation in diabetic animals [39]. Besides biochemical changes, many early or functional markers of diabetic retinopathy, nephropathy, neuropathy, and even cardiovascular disease have been reported to be prevented or reversed by antioxidants, including blood flow, nerve conduction velocity, permeability, endothelial dysfunction, albuminuria, and vascular contractility [38]. A few reports have indicated that vitamins C and E may even prevent late pathological changes in the retina and peripheral nerves of diabetic animals [39].
- PKC inhibitors may also function as antioxidants via their inhibitory effect on hyperglycemia-induced activation of NAD(P)H oxidase. These experiments only indirectly implicate oxidative stress, however, since PKC likely regulates diabetic complications on multiple levels. Oral administration of roboxistaurin (LY333531), a specific PKC-β inhibitor, to diabetic rats prevents increased albumin excretion, elevated glomerular filtration, and abnormal retinal hemodynamics [14]. Consistent with this, one report showed that angiotensin converting enzyme (ACE) inhibitors and angiotensin receptor blockers prevent the development of albuminuria in STZ-induced diabetic rats in parallel with prevention of increased expression of the p47phox component of NAD(P)H oxidase and production of its oxidative products [40].
- Human clinical trials
- Most clinical studies on the effects of antioxidant treatments have used early surrogate markers and are limited in duration and sample size.
- A small, short-term single blind study showed that administration of an antioxidant mixture (N-acetylcysteine, vitamin E, and vitamin C) to control subjects, as well as subjects with type 2 diabetes and impaired glucose tolerance reduced the increase in oxidants and endothelial markers evoked by a fatty meal [41]. Another, short-term, but randomized double blind, placebo controlled study examined the effects of vitamins E and C with or without zinc and magnesium and demonstrated reduced albumin excretion, along with evidence of reduced lipid peroxidation in groups receiving the vitamins [42]. Although many clinical trials with such antioxidant vitamins investigating the progression of diabetic complications conclude as negative or inconclusive, one may wish to see additional studies, perhaps involving a combination of these vitamins, or mega-dose therapies.
- Studies using α-lipoic acid suggest that it may improve the symptoms of diabetic polyneuropathy [13]. α-lipoic acid has the additional property of increasing glucose transport in muscle cells, which may be related to its antioxidant properties [13]. Clinical trials are now ongoing to assess the effects of PKC-β inhibition on diabetic retinopathy and neuropathy. The beneficial effects of a PKC-β specific inhibitor might also partly due to its inhibition of ROS production [14]. Further investigation into the antioxidant properties of this agent is necessary.
- Importantly, pharmacologic agents currently in use that have been shown to be effective in reducing cardiovascular mortality are known to have antioxidant properties. ACE inhibitors and HMG-CoA reductase inhibitors or statins have already demonstrated beneficial effects on diabetic subjects in large randomized controlled-trials [43]. Interestingly, ACE inhibitors, which act partially to prevent the pro-oxidant effects of angiotensin II, were shown to prevent the onset of type 2 diabetes in the Heart Outcomes Prevention Evaluation Study [43]. Statins have been shown to exert vascular antioxidative properties [44].
- Recent approaches to antioxidant therapy
- The use of new antioxidant agents that penetrate specific cellular compartments may provide a new approach to dealing with oxidative stress in diabetes. Agents such as idebenone andmito-Q, which are selectively taken up into mitochondria should be searched further [45,46]. Edaravone, 3-methyl-1-phenyl-2-pyrazolin-5-one, is a novel scavenger of free radicals [47] and has been shown to ameliorate renal ischemia/reperfusion injury by scavenging free radicals produced in renal tubular cells and inhibiting lipid peroxidation [48]. Mimetics of superoxide dismutase have been investigated in experimental models of diabetes. Tempol (4-hydroxy-2, 2, 6, 6-tetramethylpiperidine 1-oxyl) improved endothelial function in Zucker diabetic fatty rats and in db/db mice [49]. As NAD(P)H oxidase is an important source of reactive oxygen species, targeting this enzyme may provide another approach to reducing the oxidative stress of diabetes and its consequences. Indeed, administration of the NAD(P)H oxidase assembly inhibitor apocynin attenuated the long-term effects of streptozotocin-induced diabetes in producing albuminuria and glomerulosclerosis [50]. Resveratrol is a polyphenol phytoalexin that can reduce vascular superoxide levels [51] and restore endothelial function in type 2 diabetes by inhibiting tumor necrosis factor-alpha induced activation of NAD(P)H oxidase and preserving eNOS phosphorylation [52]. NF-E2-related factor-2 (Nrf2) is a transactivator of genes that contain an antioxidant response element in their promoter. Such genes code for a number of enzymes including NADPH:quinone oxidoreductase, glutathione S-transferases and aldo-keto reductases, that have an important role in the protection of cells against oxidative stress [52].
TRIALS OF ANTIOXIDANT THERAPY FOR DIABETIC VASCULAR COMPLICATIONS
- The present paper is a review of published studies on the role of ROS/RNS in the pathogenesis of diabetic vascular complications. A large number of studies indicate that oxidant production is clearly increased through multiple pathways as a result of hyperglycemia. It has recently been suggested that diabetic subjects with vascular complications may have a defective cellular antioxidant response against the oxidative and nitrosative stress generated by hyperglycemia. This raises the concept that antioxidant therapy may be of great benefit to these subjects. However, supportive evidence that antioxidants can provide beneficial effects on diabetic vascular complications in large clinical studies is lacking, largely due to the lack of a suitable antioxidant supplement. Further initiatives to study the mechanisms that cause the generation of oxidative and nitrosative stress in diabetes may lead to the discovery and evaluation of new antioxidant molecules that could inhibit the diabetes-related oxidative and nitrosative stress mechanisms.
CONCLUSIONS
-
Acknowledgements
- This work was supported by a 2-Year Research Grant of Pusan National University.
ACKNOWLEDGMENTS
- 1. The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med 1993;329:977-986. ArticlePubMed
- 2. UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 1998;352:837-853. ArticlePubMed
- 3. Ceriello A. New insights on oxidative stress and diabetic complications may lead to a "causal" antioxidant therapy. Diabetes Care 2003;26:1589-1596. ArticlePubMedPDF
- 4. Maritim AC, Sanders RA, Watkins JB 3rd. Diabetes, oxidative stress, and antioxidants: a review. J Biochem Mol Toxicol 2003;17:24-38. ArticlePubMed
- 5. Gupta S, Chough E, Daley J, Oates P, Tornheim K, Ruderman NB, Keaney JF Jr. Hyperglycemia increases endothelial superoxide that impairs smooth muscle cell Na+-K+-ATPase activity. Am J Physiol Cell Physiol 2002;282:C560-C566. ArticlePubMed
- 6. Mullarkey CJ, Edelstein D, Brownlee M. Free radical generation by early glycation products: a mechanism for accelerated atherogenesis in diabetes. Biochem Biophys Res Commun 1990;173:932-939. ArticlePubMed
- 7. Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, Giardino I, Brownlee M. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000;404:787-790. ArticlePubMedPDF
- 8. Du XL, Edelstein D, Rossetti L, Fantus IG, Goldberg H, Ziyadeh F, Wu J, Brownlee M. Hyperglycemia-induced mitochon drial superoxide overproduction activates the hexosamine pathway and induces plasminogen activator inhibitor-1 expression by increasing Sp1 glycosylation. Proc Natl Acad Sci U S A 2000;97:12222-12226. ArticlePubMedPMC
- 9. Guzik TJ, Mussa S, Gastaldi D, Sadowski J, Ratnatunga C, Pillai R, Channon KM. Mechanisms of increased vascular superoxide production in human diabetes mellitus: role of NAD(P)H oxidase and endothelial nitric oxide synthase. Circulation 2002;105:1656-1662. ArticlePubMed
- 10. Jeong HY, Son SM, Kim YK, Yun MR, Lee SM, Kim CD. Tyrosine kinase-mediated activation of NADPH oxidase enhances proliferative capacity of diabetic vascular smooth muscle cells. Life Sci 2005;76:1747-1757. ArticlePubMed
- 11. Gao L, Mann GE. Vascular NAD(P)H oxidase activation in diabetes: a double-edged sword in redox signalling. Cardiovasc Res 2009;82:9-20. ArticlePubMed
- 12. Block K, Gorin Y, Abboud HE. Subcellular localization of Nox4 and regulation in diabetes. Proc Natl Acad Sci U S A 2009;106:14385-14390. ArticlePubMedPMC
- 13. Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001;414:813-820. ArticlePubMedPDF
- 14. Inoguchi T, Li P, Umeda F, Yu HY, Kakimoto M, Imamura M, Aoki T, Etoh T, Hashimoto T, Naruse M, Sano H, Utsumi H, Nawata H. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C: dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 2000;49:1939-1945. ArticlePubMedPDF
- 15. Koya D, King GL. Protein kinase C activation and the development of diabetic complications. Diabetes 1998;47:859-866. ArticlePubMedPDF
- 16. Ceolotto G, Bevilacqua M, Papparella I, Baritono E, Franco L, Corvaja C, Mazzoni M, Semplicini A, Avogaro A. Insulin generates free radicals by an NAD(P)H, phosphatidylinositol 3'-kinase-dependent mechanism in human skin fibroblasts ex vivo. Diabetes 2004;53:1344-1351. ArticlePubMedPDF
- 17. Pitocco D, Zaccardi F, Di Stasio E, Romitelli F, Santini SA, Zuppi C, Ghirlanda G. Oxidative stress, nitric oxide, and diabetes. Rev Diabet Stud 2010;7:15-25. ArticlePubMedPMC
- 18. Cagliero E, Maiello M, Boeri D, Roy S, Lorenzi M. Increased expression of basement membrane components in human endothelial cells cultured in high glucose. J Clin Invest 1988;82:735-738. ArticlePubMedPMC
- 19. Vasquez-Vivar J, Kalyanaraman B, Martasek P, Hogg N, Masters BS, Karoui H, Tordo P, Pritchard KA Jr. Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors. Proc Natl Acad Sci U S A 1998;95:9220-9225. ArticlePubMedPMC
- 20. Kim YK, Lee MS, Son SM, Kim IJ, Lee WS, Rhim BY, Hong KW, Kim CD. Vascular NADH oxidase is involved in impaired endothelium-dependent vasodilation in OLETF rats, a model of type 2 diabetes. Diabetes 2002;51:522-527. ArticlePubMedPDF
- 21. Suzuki LA, Poot M, Gerrity RG, Bornfeldt KE. Diabetes accelerates smooth muscle accumulation in lesions of atherosclerosis: lack of direct growth-promoting effects of high glucose levels. Diabetes 2001;50:851-860. PubMed
- 22. Jeong HY, Kim CD. p22phox-derived superoxide mediates enhanced proliferative capacity of diabetic vascular smooth muscle cells. Diabetes Res Clin Pract 2004;64:1-10. ArticlePubMed
- 23. Ramana KV, Friedrich B, Srivastava S, Bhatnagar A, Srivastava SK. Activation of nuclear factor-kappaB by hyperglycemia in vascular smooth muscle cells is regulated by aldose reductase. Diabetes 2004;53:2910-2920. PubMed
- 24. Fukumoto H, Naito Z, Asano G, Aramaki T. Immunohistochemical and morphometric evaluations of coronary atherosclerotic plaques associated with myocardial infarction and diabetes mellitus. J Atheroscler Thromb 1998;5:29-35. ArticlePubMed
- 25. Kashihara N, Watanabe Y, Makino H, Wallner EI, Kanwar YS. Selective decreased de novo synthesis of glomerular proteoglycans under the influence of reactive oxygen species. Proc Natl Acad Sci U S A 1992;89:6309-6313. ArticlePubMedPMC
- 26. Etoh T, Inoguchi T, Kakimoto M, Sonoda N, Kobayashi K, Kuroda J, Sumimoto H, Nawata H. Increased expression of NAD(P)H oxidase subunits, NOX4 and p22phox, in the kidney of streptozotocin-induced diabetic rats and its reversibity by interventive insulin treatment. Diabetologia 2003;46:1428-1437. ArticlePubMedPDF
- 27. Ellis EA, Guberski DL, Somogyi-Mann M, Grant MB. Increased H2O2, vascular endothelial growth factor and receptors in the retina of the BBZ/Wor diabetic rat. Free Radic Biol Med 2000;28:91-101. ArticlePubMed
- 28. Cameron NE, Cotter MA, Archibald V, Dines KC, Maxfield EK. Anti-oxidant and pro-oxidant effects on nerve conduction velocity, endoneurial blood flow and oxygen tension in non-diabetic and streptozotocin-diabetic rats. Diabetologia 1994;37:449-459. ArticlePubMedPDF
- 29. Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev 2007;87:315-424. ArticlePubMedPMC
- 30. Pacher P, Szabo C. Role of the peroxynitrite-poly(ADP-ribose) polymerase pathway in human disease. Am J Pathol 2008;173:2-13. ArticlePubMedPMC
- 31. Molnar A, Toth A, Bagi Z, Papp Z, Edes I, Vaszily M, Galajda Z, Papp JG, Varro A, Szuts V, Lacza Z, Gero D, Szabo C. Activation of the poly(ADP-ribose) polymerase pathway in human heart failure. Mol Med 2006;12:143-152. ArticlePubMedPMCPDF
- 32. Ali TK, Matragoon S, Pillai BA, Liou GI, El-Remessy AB. Peroxynitrite mediates retinal neurodegeneration by inhibiting nerve growth factor survival signaling in experimental and human diabetes. Diabetes 2008;57:889-898. ArticlePubMedPDF
- 33. Virag L, Szabo C. The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol Rev 2002;54:375-429. ArticlePubMed
- 34. Langenstroer P, Pieper GM. Regulation of spontaneous EDRF release in diabetic rat aorta by oxygen free radicals. Am J Physiol 1992;263(1 Pt 2):H257-H265. ArticlePubMed
- 35. Du XL, Edelstein D, Dimmeler S, Ju Q, Sui C, Brownlee M. Hyperglycemia inhibits endothelial nitric oxide synthase activity by posttranslational modification at the Akt site. J Clin Invest 2001;108:1341-1348. ArticlePubMedPMC
- 36. Turan B. Role of antioxidants in redox regulation of diabetic cardiovascular complications. Curr Pharm Biotechnol 2010;11:819-836. ArticlePubMed
- 37. Folli F, Corradi D, Fanti P, Davalli A, Paez A, Giaccari A, Perego C, Muscogiuri G. The role of oxidative stress in the pathogenesis of type 2 diabetes mellitus micro- and macrovascular complications: avenues for a mechanistic-based therapeutic approach. Curr Diabetes Rev 2011;7:313-324. ArticlePubMed
- 38. Cameron NE, Cotter MA. Effects of antioxidants on nerve and vascular dysfunction in experimental diabetes. Diabetes Res Clin Pract 1999;45:137-146. ArticlePubMed
- 39. Abiko T, Abiko A, Clermont AC, Shoelson B, Horio N, Takahashi J, Adamis AP, King GL, Bursell SE. Characterization of retinal leukostasis and hemodynamics in insulin resistance and diabetes: role of oxidants and protein kinase-C activation. Diabetes 2003;52:829-837. PubMed
- 40. Onozato ML, Tojo A, Goto A, Fujita T, Wilcox CS. Oxidative stress and nitric oxide synthase in rat diabetic nephropathy: effects of ACEI and ARB. Kidney Int 2002;61:186-194. ArticlePubMed
- 41. Neri S, Signorelli SS, Torrisi B, Pulvirenti D, Mauceri B, Abate G, Ignaccolo L, Bordonaro F, Cilio D, Calvagno S, Leotta C. Effects of antioxidant supplementation on postprandial oxidative stress and endothelial dysfunction: a single-blind, 15-day clinical trial in patients with untreated type 2 diabetes, subjects with impaired glucose tolerance, and healthy controls. Clin Ther 2005;27:1764-1773. ArticlePubMed
- 42. Farvid MS, Jalali M, Siassi F, Hosseini M. Comparison of the effects of vitamins and/or mineral supplementation on glomerular and tubular dysfunction in type 2 diabetes. Diabetes Care 2005;28:2458-2464. ArticlePubMedPDF
- 43. Yusuf S, Dagenais G, Pogue J, Bosch J, Sleight P. The Heart Outcomes Prevention Evaluation Study Investigators. Vitamin E supplementation and cardiovascular events in high-risk patients. N Engl J Med 2000;342:154-160. ArticlePubMed
- 44. Mason RP, Walter MF, Jacob RF. Effects of HMG-CoA reductase inhibitors on endothelial function: role of microdomains and oxidative stress. Circulation 2004;109(21 Suppl 1):II34-II41. PubMed
- 45. Dhanasekaran A, Kotamraju S, Kalivendi SV, Matsunaga T, Shang T, Keszler A, Joseph J, Kalyanaraman B. Supplementation of endothelial cells with mitochondria-targeted antioxidants inhibit peroxide-induced mitochondrial iron uptake, oxidative damage, and apoptosis. J Biol Chem 2004;279:37575-37587. ArticlePubMed
- 46. Fatehi-Hassanabad Z, Chan CB, Furman BL. Reactive oxygen species and endothelial function in diabetes. Eur J Pharmacol 2010;636:8-17. ArticlePubMed
- 47. Higashi Y. Edaravone for the treatment of acute cerebral infarction: role of endothelium-derived nitric oxide and oxidative stress. Expert Opin Pharmacother 2009;10:323-331. ArticlePubMed
- 48. Doi K, Suzuki Y, Nakao A, Fujita T, Noiri E. Radical scavenger edaravone developed for clinical use ameliorates ischemia/reperfusion injury in rat kidney. Kidney Int 2004;65:1714-1723. ArticlePubMed
- 49. Belin de Chantemele EJ, Vessieres E, Guihot AL, Toutain B, Maquignau M, Loufrani L, Henrion D. Type 2 diabetes severely impairs structural and functional adaptation of rat resistance arteries to chronic changes in blood flow. Cardiovasc Res 2009;81:788-796. ArticlePubMed
- 50. Thallas-Bonke V, Thorpe SR, Coughlan MT, Fukami K, Yap FY, Sourris KC, Penfold SA, Bach LA, Cooper ME, Forbes JM. Inhibition of NADPH oxidase prevents advanced glycation end product-mediated damage in diabetic nephropathy through a protein kinase C-alpha-dependent pathway. Diabetes 2008;57:460-469. PubMed
- 51. Li H, Forstermann U. Resveratrol: a multifunctional compound improving endothelial function. Editorial to: "Resveratrol supplementation gender independently improves endothelial reactivity and suppresses superoxide production in healthy rats" by S. Soylemez et al. Cardiovasc Drugs Ther 2009;23:425-429. ArticlePubMedPMC
- 52. Xue M, Qian Q, Adaikalakoteswari A, Rabbani N, Babaei-Jadidi R, Thornalley PJ. Activation of NF-E2-related factor-2 reverses biochemical dysfunction of endothelial cells induced by hyperglycemia linked to vascular disease. Diabetes 2008;57:2809-2817. ArticlePubMedPMCPDF
REFERENCES
Fig. 1Induction of reactive oxygen species (ROS)/reactive nitrogen species (RNS) formation by hyperglycemia in diabetes. The major enzymes responsible for ROS generation in the vasculature include NAD(P)H oxidase, xanthine oxidase, and uncoupled nitric oxide synthase (NOS). The absence of an appropriate compensation by the endogenous antioxidant defense network and increased oxidative stress leads to the activation of stress-sensitive intracellular signaling pathways and the formation of gene products that cause cellular damage and contribute to the late complications of diabetes. AGE, advanced glycation end-products; PKC, protein kinase C; SOD, superoxide dismutase; GSH, reduced glutathione; GSSG, oxidized glutathione.
Figure & Data
References
Citations
Citations to this article as recorded by 
- Relationship of phytochemicals and antioxidant activities in Gymnema inodorum leaf extracts
Onanong Nuchuchua, Wanwisa Srinuanchai, Chaisak Chansriniyom, Uthaiwan Suttisansanee, Piya Temviriyanukul, Nitra Nuengchamnong, Uracha Ruktanonchai
Heliyon.2024; 10(1): e23175. CrossRef - The Role of Glutathione and Its Precursors in Type 2 Diabetes
Dawn Tuell, George Ford, Evan Los, William Stone
Antioxidants.2024; 13(2): 184. CrossRef - Combined toxicity of microplastics and copper on Goniopora columns
Ya-Ting Chen, De-Sing Ding, Yee Cheng Lim, Cheng-Di Dong, Shu-Ling Hsieh
Environmental Pollution.2024; 345: 123515. CrossRef - Oxidative stress, endothelial dysfunction, and N-acetylcysteine in type-2 diabetes mellitus
Xin Li, Junyong Zou, Aiping Lin, Jingshu Chi, Hong Hao, Hong Chen, Zhenguo Liu
Antioxidants & Redox Signaling.2024;[Epub] CrossRef - Acacia gum: Chemistry, properties & food applications
Deepak Mudgil, Sheweta Mudgil
Food and Humanity.2024; 2: 100264. CrossRef - High glucose-induced increasing reactive nitrogen species accumulation triggered mitochondrial dysfunction, inflammation, and apoptosis in keratinocytes
Huma Rizwan, Sonu Kumar, Golden Kumari, Arttatrana Pal
Life Sciences.2023; 312: 121208. CrossRef - Vitamin D–vitamin D receptor alleviates oxidative stress in ischemic acute kidney injury via upregulating glutathione peroxidase 3
Xueqin Wu, Shiqi Tang, Qing Dai, Bin Yi, Shikun Yang, Jian Sun, Yong Zhong, Wei Lin, Jun Liu, Yan Liu, Jianwen Wang, Jishi Liu, Qin Liao, Wei Zhang, Hao Zhang
The FASEB Journal.2023;[Epub] CrossRef - Characterization, antibacterial, antioxidant, antidiabetic, and anti-inflammatory activities of green synthesized silver nanoparticles using Phragmanthera austroarabica A. G. Mill and J. A. Nyberg extract
Dina M. Khodeer, Ali M. Nasr, Shady A. Swidan, Sarah Shabayek, Roaa M. Khinkar, Mohammed M. Aldurdunji, Maryam A. Ramadan, Jihan M. Badr
Frontiers in Microbiology.2023;[Epub] CrossRef - Toxic Effects of Copper Sulfate and Trichlorfon on the Intestines of Zebrafish (Danio rerio)
Qing Li, Yangyang Cao, Yuting Wang, Xiaodong Long, Wenbo Sun, Hui Yang, Yingying Zhang, Zhitao Qi
Aquaculture Research.2023; 2023: 1. CrossRef - Early Detection Is the Best Prevention—Characterization of Oxidative Stress in Diabetes Mellitus and Its Consequences on the Cardiovascular System
Sanela Rajlic, Hendrik Treede, Thomas Münzel, Andreas Daiber, Georg Daniel Duerr
Cells.2023; 12(4): 583. CrossRef - In vitroand computational studies of the antioxidant and anti-diabetic properties ofBridelia ferruginea
Olajumoke Oyebode, Ochuko Lucky Erukainure, Lindiwe Zuma, Collins U. Ibeji, Neil Anthony Koorbanally, Md. Shahidul Islam
Journal of Biomolecular Structure and Dynamics.2022; 40(9): 3989. CrossRef - Oxidative distress in aging and age-related diseases: Spatiotemporal dysregulation of protein oxidation and degradation
Sergey Zavadskiy, Susanna Sologova, Nurbubu Moldogazieva
Biochimie.2022; 195: 114. CrossRef - Effects of Smoking on Diabetic Nephropathy
Yasemin Gündoğdu, İnan Anaforoğlu
Frontiers in Clinical Diabetes and Healthcare.2022;[Epub] CrossRef - Endogenous Protective Factors and Potential Therapeutic Agents for Diabetes-Associated Atherosclerosis
Chaoqun Wang, Jin Chen, Pin Wang, Shengli Qing, Wenwen Li, Jin Lu
Frontiers in Endocrinology.2022;[Epub] CrossRef - Antihyperglycemic activity of verbenone and L-arginine in nicotinamide-streptozotocin-induced diabetic mice: in vitro and in vivo studies
Habibu Tijjani, Abdulkadir Mohammed Danyaro, Ahmed Olatunde, Aminu Umar Kura
Beni-Suef University Journal of Basic and Applied Sciences.2022;[Epub] CrossRef - Comparison of the combined toxicity of polystyrene microplastics and different concentrations of cadmium in zebrafish
Hui Yang, Zhu Zhu, Yuexuan Xie, Chen Zheng, Zhenyuan Zhou, Tianhao Zhu, Yingying Zhang
Aquatic Toxicology.2022; 250: 106259. CrossRef - “EVALUATION OF HYPERGLYCEMIA INDUCED OXIDATIVE STRESS IN TYPE-2 DIABETIC PATIENT”
Ameerul Hasan Amir, Faisal Iqubal, Savita Rathor, Afreena Nasir
INTERNATIONAL JOURNAL OF SCIENTIFIC RESEARCH.2022; : 12. CrossRef - Evaluation of Anti-Hyperglycemia and Complications of Red and Black Thai Jasmine Rice Cultivars in Streptozotocin-Induced Diabetic Rats
Nittiya Suwannasom, Chutamas Thepmalee, Krissana Khoothiam, Chonthida Thephinlap
Molecules.2022; 27(22): 8043. CrossRef - p38 MAPK Inhibitor (SB203580) and Metformin Reduces Aortic Protein Carbonyl and Inflammation in Non-obese Type 2 Diabetic Rats
Nuttikarn Nokkaew, Podsawee Mongkolpathumrat, Ruttanapong Junsiri, Supawit Jindaluang, Nichagron Tualamun, Niya Manphatthanakan, Nareumon Saleesee, Marisa Intasang, Jantira Sanit, Punyanuch Adulyaritthikul, Kantapich Kongpol, Sarawut Kumphune, Nitirut Ner
Indian Journal of Clinical Biochemistry.2021; 36(2): 228. CrossRef - Elucidation of phytochemicals and antioxidants properties of Sasa quelpaertensis
Hee Chul Ko, Mi Gyeong Jang, Jae-Won Kim, Songyee Baek, Nam Ho Lee, Se-Jae Kim
International Journal of Food Properties.2021; 24(1): 210. CrossRef - Organelle dynamics of endothelial mitochondria in diabetic angiopathy
Hong Xiang, Ruipeng Song, Jie Ouyang, Ruifang Zhu, Zhihao Shu, Yulan Liu, Xuewen Wang, Dongtao Zhang, Jiangwei Zhao, Hongwei Lu
European Journal of Pharmacology.2021; 895: 173865. CrossRef -
Hypoglycemic and antioxidant effects of green tomato (
Physalis ixocarpa
Brot.) calyxes’ extracts
Fernando Guerrero‐Romero, Luis E. Simental‐Mendía, María Inés Guerra Rosas, Víctor Iván Sayago‐Monreal, Juliana Morales Castro, Claudia I. Gamboa‐Gómez
Journal of Food Biochemistry.2021;[Epub] CrossRef - Anti-anaemic effect of ethanol leaf extract of Cnidosculus aconitifolius on cyclophosphomide-induced anaemia in rats
Jamila Abdulhamid Atata, Taiwo Oluwaseun Ayoola, Abdullateef Abiodun Ajadi, Sani Adamu, Aishat O. Olatunji, Khalid Talha Biobaku
Journal of Complementary and Integrative Medicine.2021; 18(1): 87. CrossRef - NLRP3 Inflammasome at the Interface of Inflammation, Endothelial Dysfunction, and Type 2 Diabetes
Ilona M. Gora, Anna Ciechanowska, Piotr Ladyzynski
Cells.2021; 10(2): 314. CrossRef - Potential Roles of Endoplasmic Reticulum Stress and Cellular Proteins Implicated in Diabesity
Sagir Mustapha, Mustapha Mohammed, Ahmad Khusairi Azemi, Ismaeel Yunusa, Aishatu Shehu, Lukman Mustapha, Yusuf Wada, Mubarak Hussaini Ahmad, Wan Amir Nizam Wan Ahmad, Aida Hanum Ghulam Rasool, Siti Safiah Mokhtar, Silvana Hrelia
Oxidative Medicine and Cellular Longevity.2021; 2021: 1. CrossRef - Inflammation and apoptosis, two key events induced by hyperglycemia mediated reactive nitrogen species in RGC-5 cells
Sweta Pal, G. Nageswar Rao, Arttatrana Pal
Life Sciences.2021; 279: 119693. CrossRef - The Predictive Value of Neutrophil-to-Lymphocyte Ratio and Platelet-to-Lymphocyte Ratio Levels of Diabetic Peripheral Neuropathy
Meiqiao Chen, Yuyou Zhu, Jumei Wang, Guoping Wang, Yuanbo Wu
Journal of Pain Research.2021; Volume 14: 2049. CrossRef - The Role of Endoplasmic Reticulum Stress and NLRP3 Inflammasomes in the Development of Atherosclerosis
V. V. Pushkarev, L. K. Sokolova, O. I. Kovzun, V. M. Pushkarev, M. D. Tronko
Cytology and Genetics.2021; 55(4): 331. CrossRef - Aging under Pressure: The Roles of Reactive Oxygen and Nitrogen Species (RONS) Production and Aging Skeletal Muscle in Endothelial Function and Hypertension—From Biological Processes to Potential Interventions
Hollie Speer, Andrew J. McKune
Antioxidants.2021; 10(8): 1247. CrossRef - Natural Antioxidants in New Age-Related Diseases
Arti Devi, Vagish Dwibedi, Zaved Ahmed Khan
Revista Brasileira de Farmacognosia.2021; 31(4): 387. CrossRef - Potential risks of endoplasmic reticulum stress on vasculopathy in diabetes
Sagir Mustapha, Mustapha Mohammed, Ismaeel Yunusa, Aida Hanum Ghulam Rasool, Siti Safiah Mokhtar
Obesity Medicine.2020; 19: 100274. CrossRef - Potential roles of Citrulline and watermelon extract on metabolic and inflammatory variables in diabetes mellitus, current evidence and future directions: A systematic review
Samaneh Azizi, Reza Mahdavi, Elnaz Vaghef‐Mehrabany, Vahid Maleki, Nahid Karamzad, Mehrangiz Ebrahimi‐Mameghani
Clinical and Experimental Pharmacology and Physiology.2020; 47(2): 187. CrossRef - Enzymic polypeptide antioxidant activity and inhibitory activity on α-glucosidase and α-amylase from Paeonia ostii cake
Huiru Qiao, Xiaojuan Bi, Yuanyuan Zhang, Mengran Liu, Shuchong Zu, Nan Jia, Shougang Jiang, Qi Lu, Yuangang Zu, Yihong Bao
Industrial Crops and Products.2020; 146: 112158. CrossRef -
Hypoglycemic and antioxidant properties of konjac (
Amorphophallus konjac
) in vitro and in vivo
Claudia I. Gamboa‐Gómez, Fernando Guerrero‐Romero, Miguel A. Sánchez‐Meraz, Luis E. Simental‐Mendía
Journal of Food Biochemistry.2020;[Epub] CrossRef - Heme on Pulmonary Malaria: Friend or Foe?
Tatiana Almeida Pádua, Mariana Conceição Souza
Frontiers in Immunology.2020;[Epub] CrossRef - The role of reactive oxygen species in the pathogenesis and treatment of retinal diseases
Thomas CW. Chan, Jennifer L. Wilkinson Berka, Devy Deliyanti, Damien Hunter, Adrian Fung, Gerald Liew, Andrew White
Experimental Eye Research.2020; 201: 108255. CrossRef - The Effect of an Atherogenic Diet and Acute Hyperglycaemia on Endothelial Function in Rabbits Is Artery Specific
Alexander Tacey, Tawar Qaradakhi, Cassandra Smith, Chris Pittappillil, Alan Hayes, Anthony Zulli, Itamar Levinger
Nutrients.2020; 12(7): 2108. CrossRef - Cathelicidin Modulates Vascular Smooth Muscle Cell Phenotypic Switching through ROS/IL-6 Pathway
Xiaoliang Dong, Di Wu, Yihan Zhang, Lingling Jia, Xiaohua Pan, Jia Sun, Li-Long Pan
Antioxidants.2020; 9(6): 491. CrossRef - Immune-Inflammation in Atherosclerosis: A New Twist in an Old Tale
Atefe Ghamar Talepoor, Hamed Fouladseresht, Shahdad Khosropanah, Mehrnoosh Doroudchi
Endocrine, Metabolic & Immune Disorders - Drug Targets.2020; 20(4): 525. CrossRef - Fabrication of sensitive D-fructose sensor based on facile ternary mixed ZnO/CdO/SnO2 nanocomposites by electrochemical approach
M.M. Alam, Abdullah M. Asiri, Mohammed M. Rahman, M.A. Islam
Surfaces and Interfaces.2020; 19: 100540. CrossRef - The role of glutathione and glutathione peroxidase in regulating cellular level of reactive oxygen and nitrogen species
Sheetal Panday, Raghav Talreja, Mahendra Kavdia
Microvascular Research.2020; 131: 104010. CrossRef - Evaluation of the Antioxidant and Antidiabetic Potential of the Poly Herbal Formulation: Identification of Bioactive Factors
V.V. Sathibabu Uddandrao, Parim Brahmanaidu, Saravanan Ganapathy
Cardiovascular & Hematological Agents in Medicinal Chemistry.2020; 18(2): 111. CrossRef - Rule of UA on Cardiac Myocytes Uric Acid Differently Influence the Oxidative Damage Induced by Acute Exposure of High Level of Glucose in Chicken Cardiac Myocytes
Xiaolong Sun, Hongchao Jiao, Jingpeng Zhao, Xiaojuan Wang, Hai Lin
Frontiers in Veterinary Science.2020;[Epub] CrossRef - Elevated Serum Levels of Ischemia Modified Albumin and Malondialdehyde are Related to Atherogenic Index of Plasma in a Cohort of Prediabetes
Mervat M. El-Eshmawy, Doaa F. Gad, Azza A. El-Baiomy
Endocrine, Metabolic & Immune Disorders - Drug Targets.2020; 20(8): 1347. CrossRef - Low serum bilirubin levels contribute to the presence and progression of distal symmetrical polyneuropathy in Chinese patients with type 2 diabetes
J. Jin, W. Wang, T. Gu, C. Chen, J. Sun, W. Chen, Y. Bi, D. Zhu
Diabetes & Metabolism.2019; 45(1): 47. CrossRef - Therapeutic Benefit of Dillenia indica in Diabetes and Its Associated Complications
Parul Kamboj, Narayan C. Talukdar, Sanjay K. Banerjee
Journal of Diabetes Research.2019; 2019: 1. CrossRef - The impact of xanthine oxidase (XO) on hemolytic diseases
Heidi M. Schmidt, Eric E. Kelley, Adam C. Straub
Redox Biology.2019; 21: 101072. CrossRef - What is responsible for antioxidant properties of polyphenolic compounds from plants?
Małgorzata Olszowy
Plant Physiology and Biochemistry.2019; 144: 135. CrossRef - Leucocyte Telomere Length and Glucose Tolerance Status in Mixed-Ancestry South Africans
Cecil J. Weale, Glenda M. Davison, Gloudina M. Hon, Andre P. Kengne, Rajiv T. Erasmus, Tandi E. Matsha
Cells.2019; 8(5): 464. CrossRef - Chronic Inhibition of Mitochondrial Dihydrolipoamide Dehydrogenase (DLDH) as an Approach to Managing Diabetic Oxidative Stress
Xiaojuan Yang, Jing Song, Liang-Jun Yan
Antioxidants.2019; 8(2): 32. CrossRef - Cilostazol Attenuates Retinal Oxidative Stress and Inflammation in a Streptozotocin-Induced Diabetic Animal Model
Po-Ting Yeh, Yu-Hsun Huang, Shu-Wen Chang, Lu-Chun Wang, Chung-May Yang, Wei-Shiung Yang, Chung-Wu Lin, Chang-Hao Yang
Current Eye Research.2019; 44(3): 294. CrossRef - Crosstalk Between Oxidative Stress and Endoplasmic Reticulum (ER) Stress in Endothelial Dysfunction and Aberrant Angiogenesis Associated With Diabetes: A Focus on the Protective Roles of Heme Oxygenase (HO)-1
Hatem Maamoun, Tarek Benameur, Gianfranco Pintus, Shankar Munusamy, Abdelali Agouni
Frontiers in Physiology.2019;[Epub] CrossRef - Novel Curcumin C66 That Protects Diabetes-Induced Aortic Damage Was Associated with Suppressing JNK2 and Upregulating Nrf2 Expression and Function
Cheng Li, Xiao Miao, Shudong Wang, Binay Kumar Adhikari, Xin Wang, Jian Sun, Quan Liu, Qian Tong, Yonggang Wang
Oxidative Medicine and Cellular Longevity.2018; 2018: 1. CrossRef - Nebivolol, a β-blocker abrogates streptozotocin-induced behavioral, biochemical, and neurophysiological deficit by attenuating oxidative-nitrosative stress: a possible target for the prevention of diabetic neuropathy
Naini Bhadri, Rema Razdan, Sumanta Kumar Goswami
Naunyn-Schmiedeberg's Archives of Pharmacology.2018; 391(2): 207. CrossRef - Angiogenic Dysregulation in Pregnancy-Related Hypertension—A Role for Metformin
Nerolen Soobryan, Saravanakumar Murugesan, Arunagiri Pandiyan, Jagidesa Moodley, Irene Mackraj
Reproductive Sciences.2018; 25(11): 1531. CrossRef - Nutrients and Oxidative Stress: Friend or Foe?
Bee Ling Tan, Mohd Esa Norhaizan, Winnie-Pui-Pui Liew
Oxidative Medicine and Cellular Longevity.2018; 2018: 1. CrossRef - Free radical scavenging, α-glucosidase inhibitory and lipase inhibitory activities of eighteen Sudanese medicinal plants
Sara Mustafa Idris Elbashir, Hari Prasad Devkota, Mikiyo Wada, Naoki Kishimoto, Masataka Moriuchi, Tsuyoshi Shuto, Shogo Misumi, Hirofumi Kai, Takashi Watanabe
BMC Complementary and Alternative Medicine.2018;[Epub] CrossRef - An Update on Hypertension in Children With Type 1 Diabetes
Mallory L. Downie, Emma H. Ulrich, Damien G. Noone
Canadian Journal of Diabetes.2018; 42(2): 199. CrossRef - Chronic Endurance Exercise Impairs Cardiac Structure and Function in Middle-Aged Mice with Impaired Nrf2 Signaling
Gobinath Shanmugam, Madhusudhanan Narasimhan, Robbie L. Conley, Thiagarajan Sairam, Ashutosh Kumar, Ronald P. Mason, Ramalingam Sankaran, John R. Hoidal, Namakkal S. Rajasekaran
Frontiers in Physiology.2017;[Epub] CrossRef - Protective effects of 6-Gingerol on vascular endothelial cell injury induced by high glucose via activation of PI3K-AKT-eNOS pathway in human umbilical vein endothelial cells
Dan Liu, Mengqing Wu, Yi Lu, Tao Xian, Yupeng Wang, Bowei Huang, Guohua Zeng, Qiren Huang
Biomedicine & Pharmacotherapy.2017; 93: 788. CrossRef - Evaluation of the release profile, stability and antioxidant activity of a proanthocyanidin-rich cinnamon (Cinnamomum zeylanicum) extract co-encapsulated with α-tocopherol by spray chilling
Fabrício L. Tulini, Volnei B. Souza, Marcelo Thomazini, Marluci P. Silva, Adna P. Massarioli, Severino M. Alencar, Eliria M.J.A. Pallone, Maria I. Genovese, Carmen S. Favaro-Trindade
Food Research International.2017; 95: 117. CrossRef - Modulatory Effect of Achyranthes aspera L., Seeds on Carbohydrates and Protein Bound Enzymes on High Fructose Fed Diet
Veerappan Ramanathan, Malarvili Thekkumala
Journal of Pharmacology and Toxicology.2017; 12(3): 154. CrossRef - Pyrethroid insecticide lambda-cyhalothrin and its metabolites induce liver injury through the activation of oxidative stress and proinflammatory gene expression in rats following acute and subchronic exposure
Bakhta Aouey, Mohamed Derbali, Yassine Chtourou, Michèle Bouchard, Abdelmajid Khabir, Hamadi Fetoui
Environmental Science and Pollution Research.2017; 24(6): 5841. CrossRef - Effect of hydroxychloroquine on oxidative/nitrosative status and angiogenesis in endothelial cells under high glucose condition
Aysa Rezabakhsh, Soheila Montazersaheb, Elahe Nabat, Mehdi Hassanpour, Azadeh Montaseri, Hassan Malekinejad, Ali Akbar Movassaghpour, Reza Rahbarghazi, Alireza Garjani
BioImpacts.2017; 7(4): 219. CrossRef - The Effect of Vitamin E on Oxidative Stress Indicated by Serum Malondialdehyde in Insulin-dependent Type 2 Diabetes Mellitus Patients with Retinopathy
Irini P. Chatziralli, George Theodossiadis, Prodromos Dimitriadis, Michail Charalambidis, Antonios Agorastos, Zisis Migkos, Nikolaos Platogiannis, Marilita M. Moschos, Panagiotis Theodossiadis, Petros Keryttopoulos
The Open Ophthalmology Journal.2017; 11(1): 51. CrossRef - Association between Fluorescent Advanced Glycation End-Products and Vascular Complications in Type 2 Diabetic Patients
Alexis Guerin-Dubourg, Maxime Cournot, Cynthia Planesse, Xavier Debussche, Olivier Meilhac, Philippe Rondeau, Emmanuel Bourdon
BioMed Research International.2017; 2017: 1. CrossRef - Hyperglycemic Stress and Carbon Stress in Diabetic Glucotoxicity
Xiaoting Luo, Jinzi Wu, Siqun Jing, Liang-Jun Yan
Aging and disease.2016; 7(1): 90. CrossRef - İNSAN VE HAYVANLARDA DİABETES MELLİTUS
Özlem ÖZMEN, Senay TOPSAKAL
Mehmet Akif Ersoy Üniversitesi Veteriner Fakültesi Dergisi.2016; 1(1): 47. CrossRef - The novel mitochondria-targeted hydrogen sulfide (H 2 S) donors AP123 and AP39 protect against hyperglycemic injury in microvascular endothelial cells in vitro
Domokos Gerő, Roberta Torregrossa, Alexis Perry, Alicia Waters, Sophie Le-Trionnaire, Jacqueline L. Whatmore, Mark Wood, Matthew Whiteman
Pharmacological Research.2016; 113: 186. CrossRef - Heated apple juice supplemented with onion has greatly improved nutritional quality and browning index
Bonggi Lee, Jeong Dae Seo, Jin-Kyu Rhee, Choon Young Kim
Food Chemistry.2016; 201: 315. CrossRef - Inverse association between serum total bilirubin levels and diabetic peripheral neuropathy in patients with type 2 diabetes
Eun Sook Kim, Sung Won Lee, Eun Young Mo, Sung Dae Moon, Je Ho Han
Endocrine.2015; 50(2): 405. CrossRef - Interactions of short-acting, intermediate-acting and pre-mixed human insulins with free radicals—Comparative EPR examination
Paweł Olczyk, Katarzyna Komosinska-Vassev, Paweł Ramos, Łukasz Mencner, Krystyna Olczyk, Barbara Pilawa
International Journal of Pharmaceutics.2015; 490(1-2): 9. CrossRef - Long-term high fat feeding of rats results in increased numbers of circulating microvesicles with pro-inflammatory effects on endothelial cells
L. F. Heinrich, D. K. Andersen, M. E. Cleasby, C. Lawson
British Journal of Nutrition.2015; 113(11): 1704. CrossRef - Gum Arabic extracts protect against hepatic oxidative stress in alloxan induced diabetes in rats
Abdelkareem A. Ahmed, Jaafar S. Fedail, Hassan H. Musa, Asghar Ali Kamboh, Amal Z. Sifaldin, Taha H. Musa
Pathophysiology.2015; 22(4): 189. CrossRef - Reduced muscarinic parotid secretion is underlain by impaired NO signaling in diabetic rabbits
J Roganović, LJ Djukić, E Kršljak, N Tanić, D Stojić
Oral Diseases.2015; 21(5): 634. CrossRef - Polycomb Repressive Complex 2 Regulates MiR-200b in Retinal Endothelial Cells: Potential Relevance in Diabetic Retinopathy
Michael Anthony Ruiz, Biao Feng, Subrata Chakrabarti, Rong Wen
PLOS ONE.2015; 10(4): e0123987. CrossRef - Anti-hyperglycaemic effect ofPhragmenthera austroarabicaA.G.Mill. & J.A.Nyberg extract in streptozotocin-induced diabetes in rats
Abeer Hanafy, Jihan M. Badr
Natural Product Research.2014; 28(24): 2351. CrossRef - Functional and morphological changes of the diencephalon vascular endothelium of rats in experimental ischemia-reperfusion injury and diabetes mellitus
TM Boĭchuk, TP Savchuk
Fiziolohichnyĭ zhurnal.2014; 59(6): 30. CrossRef - Neovascularization in diabetes and its complications. Unraveling the angiogenic paradox
Paulo Zoé Costa, Raquel Soares
Life Sciences.2013; 92(22): 1037. CrossRef - Oxidative-stress-induced epigenetic changes in chronic diabetic complications
Biao Feng, Michael Anthony Ruiz, Subrata Chakrabarti
Canadian Journal of Physiology and Pharmacology.2013; 91(3): 213. CrossRef - Effect of α-Linolenic Acid on Streptozotocin-induced Diabetic Retinopathy Indices In Vivo
Jun-hui Shen, Qi Ma, Shen-grong Shen, Guo-Tong Xu, Undurti N. Das
Archives of Medical Research.2013; 44(7): 514. CrossRef - Antihyperglycemic Effect ofGinkgo bilobaExtract in Streptozotocin-Induced Diabetes in Rats
Daye Cheng, Bin Liang, Yunhui Li
BioMed Research International.2013; 2013: 1. CrossRef
 PubReader
PubReader Cite
Cite