- Current
- Browse
- Collections
-
For contributors
- For Authors
- Instructions to authors
- Article processing charge
- e-submission
- For Reviewers
- Instructions for reviewers
- How to become a reviewer
- Best reviewers
- For Readers
- Readership
- Subscription
- Permission guidelines
- About
- Editorial policy
Articles
- Page Path
- HOME > Diabetes Metab J > Volume 45(2); 2021 > Article
-
ReviewBasic Research Application of Animal Models in Diabetic Cardiomyopathy
-
Wang-Soo Lee1
 , Jaetaek Kim2
, Jaetaek Kim2 -
Diabetes & Metabolism Journal 2021;45(2):129-145.
DOI: https://doi.org/10.4093/dmj.2020.0285
Published online: March 25, 2021
1Division of Cardiology, Department of Internal Medicine, Chung-Ang University College of Medicine, Seoul, Korea
2Division of Endocrinology and Metabolism, Department of Internal Medicine, Chung-Ang University College of Medicine, Seoul, Korea
-
Corresponding authors: Wang-Soo Lee Division of Cardiology, Department of Internal Medicine, Chung-Ang University Hospital, 102 Heukseok-ro, Dongjak-gu, Seoul 06973, Korea E-mail: wslee1227@cau.ac.kr
-
Jaetaek Kim Division of Endocrinology and Metabolism, Department of Internal Medicine, ChungAng University Hospital, 102 Heukseok-ro, Dongjak-gu, Seoul 06973, Korea E-mail: jtkim@cau.ac.kr
Copyright © 2021 Korean Diabetes Association
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- ABSTRACT
- Graphical abstract
- INTRODUCTION
- PATHOGENESIS OF DIABETIC CARDIOMYOPATHY
- ANIMAL MODELS ACCORDING TO PATHOMECHANISMS OF DIABETIC CARDIOMYOPATHY
- ANIMAL MODELS FOR HEART FAILURE WITH PRESERVED EJECTION FRACTION
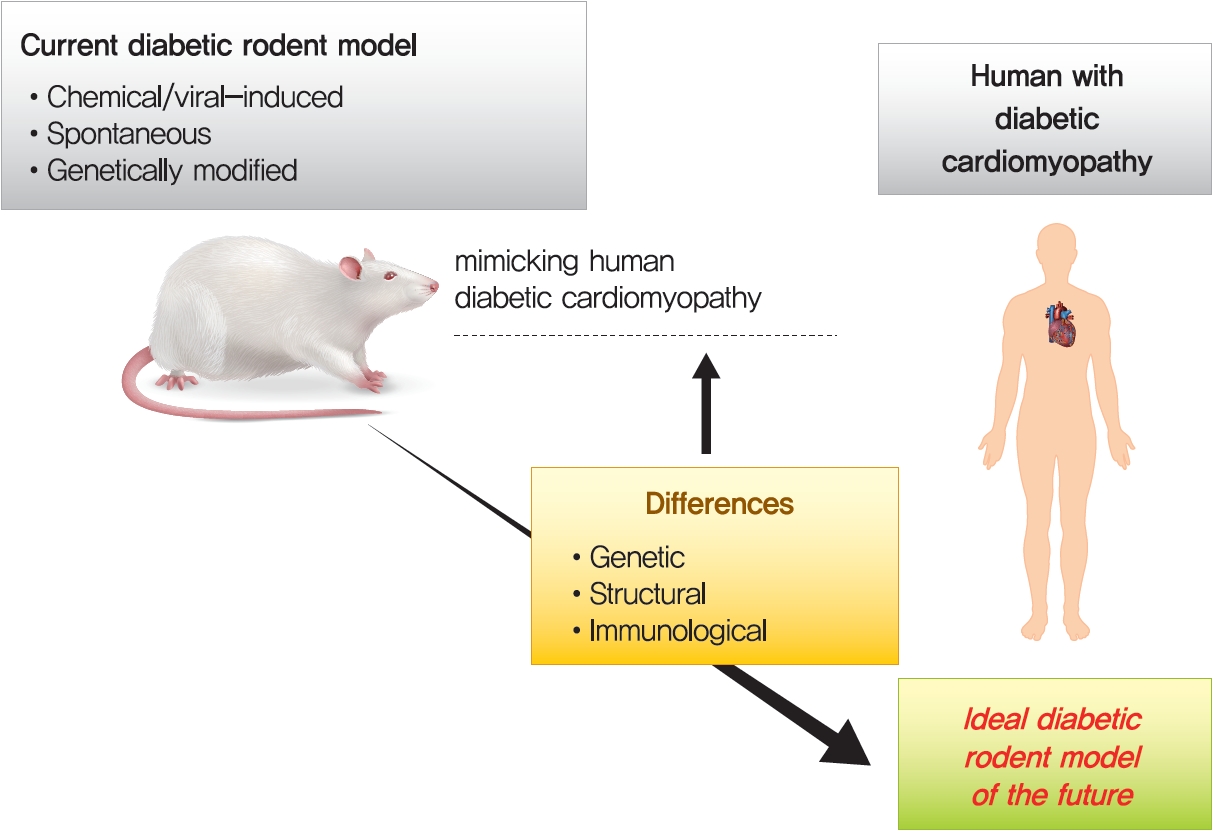
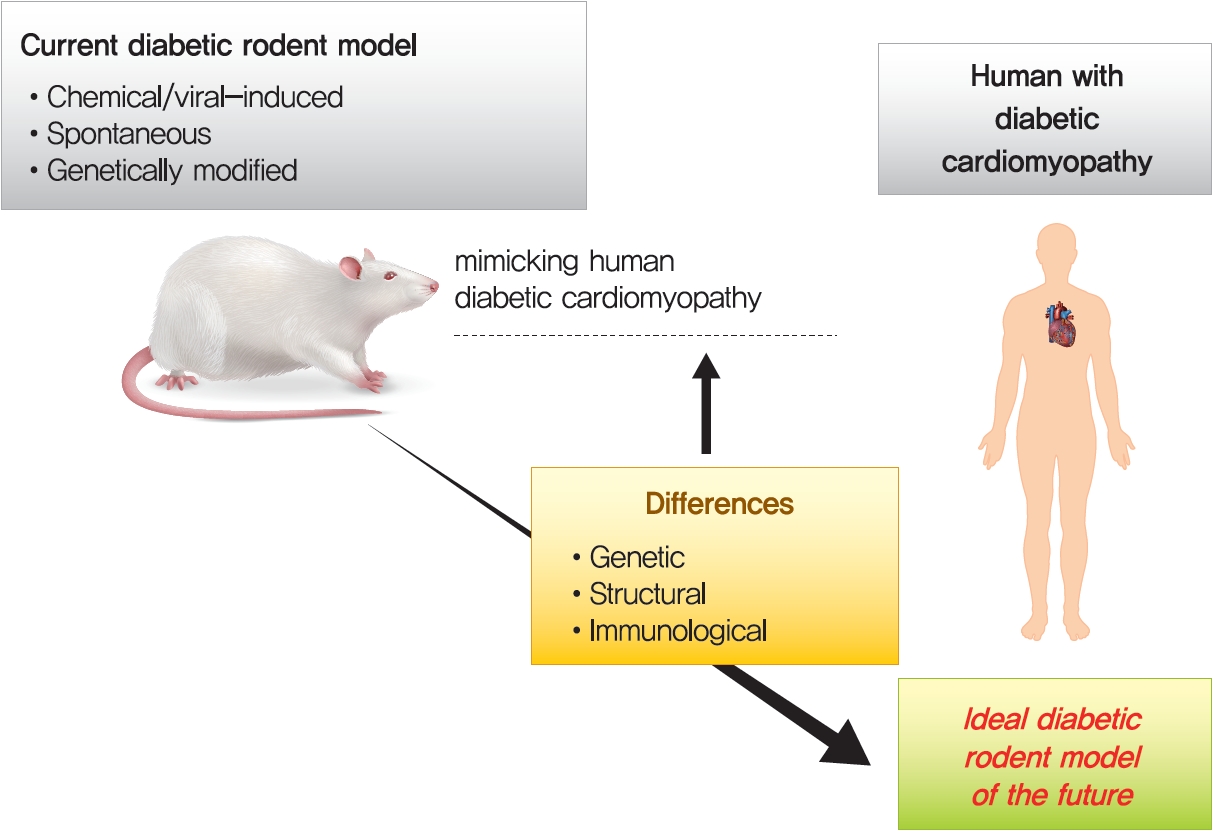
- THE DIFFERENCES BETWEEN HUMAN PATIENTS AND ANIMAL MODELS IN DIABETIC CARDIOMYOPATHY
- SUMMARY AND PERSPECTIVES
- NOTES
- REFERENCES
ABSTRACT
- Diabetic heart disease is a growing and important public health risk. Apart from the risk of coronary artery disease or hypertension, diabetes mellitus (DM) is a well-known risk factor for heart failure in the form of diabetic cardiomyopathy (DiaCM). Currently, DiaCM is defined as myocardial dysfunction in patients with DM in the absence of coronary artery disease and hypertension. The underlying pathomechanism of DiaCM is partially understood, but accumulating evidence suggests that metabolic derangements, oxidative stress, increased myocardial fibrosis and hypertrophy, inflammation, enhanced apoptosis, impaired intracellular calcium handling, activation of the renin-angiotensin-aldosterone system, mitochondrial dysfunction, and dysregulation of microRNAs, among other factors, are involved. Numerous animal models have been used to investigate the pathomechanisms of DiaCM. Despite some limitations, animal models for DiaCM have greatly advanced our understanding of pathomechanisms and have helped in the development of successful disease management strategies. In this review, we summarize the current pathomechanisms of DiaCM and provide animal models for DiaCM according to its pathomechanisms, which may contribute to broadening our understanding of the underlying mechanisms and facilitating the identification of possible new therapeutic targets.
- The prevalence of diabetes mellitus (DM) is increasing at a critical rate; recent assumptions predict that 642 million adults worldwide will be affected by DM by 2040 [1,2]. Importantly, diabetic patients have an increased risk of chronic complications, including retinopathy, neuropathy, nephropathy, and cardiovascular disease [1,3,4].
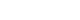
- The Framingham Heart Study revealed that the risk of heart failure (HF) increases 2- to 8-fold in the presence of type 2 diabetes mellitus (T2DM) and that 19% of patients with HF have T2DM [5,6]. In fact, patients with diabetes can develop a unique form of HF, termed diabetic cardiomyopathy (DiaCM), which is characterized by initial diastolic dysfunction without systolic dysfunction, often referred to as HF with preserved ejection fraction (HFpEF), eventually progressing to HF with reduced ejection fraction [7,8]. DM elicits changes in several cell types in the heart, including cardiac fibroblasts, endothelial cells, cardiomyocytes, and inflammatory cells. These changes promote detrimental cardiac remodeling, including cardiac fibrosis, cardiomyocyte apoptosis, and myocardial hypertrophy [1,9,10].
- Many animal models of chronic hyperglycemia exist, each replicating certain aspects of clinical DM. These animal models use genetic engineering, obesogenic diets and pancreatic toxins to induce DM. In terms of DiaCM, several models of DM have been shown to cause diastolic dysfunction [1]. Despite these efforts, effective treatment options have remained elusive, partly due to the limitations of an experimental model that adequately mimics human DiaCM [1].
- This review provides an overview of the pathomechanisms of DiaCM. We also describe the small animal models for DiaCM according to its pathomechanisms. These findings will aid our understanding of the pathophysiology of DiaCM and hopefully advance the discovery of new therapeutic strategies for this unique disease entity.
INTRODUCTION
- The pathomechanisms underlying the development of DiaCM are multifactorial and incompletely understood. There are various proposed mechanisms of DiaCM, including metabolic disturbances, insulin resistance, cardiac autonomic dysfunction, maladaptive immune responses, subcellular component abnormalities, microvascular impairment, and alterations in the renin-angiotensin-aldosterone system (RAAS) [5,11,12]. These factors induce the activation of multiple inflammatory pathways and increase oxidative stress, which mediate extracellular and cellular injuries, thus ultimately inducing pathological cardiac remodeling [5,13].
PATHOGENESIS OF DIABETIC CARDIOMYOPATHY
- Rodents, especially rats and mice, are powerful tools to investigate the pathophysiological mechanisms involved in the development of DiaCM. Rat or mouse genomes are approximately the same size as the human genome, each containing nearly 30,000 protein-coding genes, with approximately 99% of the genes encoded in the mouse genome having a homologue in humans [14-16]. In addition to these genomic resemblances, further benefits of mouse models include the short breeding cycle and the usefulness of a variety of genetically engineered loss- and gain-of-function models [14,17]. The commonly used rodent models to produce type 1 diabetes mellitus (T1DM) and T2DM are summarized in Table 1 [14,17-21] and Fig. 1. The following sections will describe the animal models according to the pathomechanisms of DiaCM observed in T1DM and T2DM.
- Metabolic derangements
- Innumerable studies apply dietary manipulations to induce obesity, insulin resistance, and T2DM in rodents and large animal models [14,17,19]. Insulin signaling in the heart is preserved in T2DM rodent models following short-term high-fat diet (HFD) feeding [22,23]. However, prolonged HFD feeding in animal models impairs its downstream targets of the serine/threonine kinase Akt and forkhead box O-1 (FOXO1) transcription factor phosphorylation [24], which results in persistent FOXO1 nuclear localization and activation. Mice with cardiac-specific deletion of glucose transporter type 4 (GLUT4) showed normal cardiac function in the unstressed state but developed maladaptive hypertrophy and severe contractile dysfunction in response to left ventricular (LV) pressure overload [13,25]. Therefore, GLUT4 is required for the maintenance of cardiac function and structure in response to pathological processes that increase energy demand, in part through secondary changes in mitochondrial metabolism and cellular stress survival signaling, such as the phosphoinositide 3-kinase (PI3K)–Akt pathway [13,25].
- In addition to stimulating glucose uptake, both insulin signaling [13,26] and cardiomyocyte contraction [13,27] can promote fatty acid uptake into cardiomyocytes via induction of cluster of differentiation 36 (CD36) translocation to sarcolemma membranes [26,28]. The long-lasting presence of CD36 at the sarcolemma membrane leads to an increased rate of long-chain fatty acid uptake and accumulation of triglycerides in cardiomyocytes, which results in lipotoxic DiaCM [28,29].
- The transcription factor peroxisome proliferator-activated receptor-α (PPARα) is a major regulator of lipid metabolism and can increase the expression of genes encoding CD36, fatty acid-binding proteins and proteins involved in β-oxidation in the mitochondria and peroxisome [13,30]. Tribbles-related protein 3 (TRB3) can directly bind to Akt and inhibit Akt phosphorylation [13,31,32]. The expression of TRB3 is upregulated in the heart in T1DM and T2DM rodent models [33,34] and in skeletal muscle in patients with T2DM [32]. Furthermore, a rat model of T2DM induced by a HFD and low-dose streptozotocin (STZ) demonstrated severe insulin resistance and properties of DiaCM, including myocardial fibrosis, cardiac inflammation and LV dysfunction, in addition to increased expression of TRB3, compared with control rats [34].
- The hearts from rats with T2DM infused ex vivo with the CD36 inhibitor sulfo-N-succinimidyl oleate (SSO) before inducing hypoxia, which resulted in a 29% reduction in the rate of fatty acid oxidation and an approximately 50% reduction in triglyceride concentration compared with vehicle treatment, showed a restoration of fatty acid metabolism to control levels following hypoxia–reoxygenation [13,35]. SSO infusion into diabetic rat hearts ex vivo before hypoxia also prevented cardiac dysfunction [35]. Fenofibrate treatment prevented fibrosis and diastolic dysfunction in diabetic rats, probably through improvements in cardiac and systemic lipid metabolism [36,37]. Fenofibrate treatment was also associated with reductions in markers of apoptosis and cardiac hypertrophy in rats with STZ-induced T1DM [38]. The glucagon-like peptide-1 (GLP1) analog liraglutide protected against the development of DiaCM in a rat model of STZ-induced T1DM by inhibiting the endoplasmic reticulum (ER) stress pathway [39]. Similarly, the GLP1 analog exendin-4 prevented the development of DiaCM via the amelioration of lipotoxicity in a mouse model of T2DM [40]. The dipeptidyl peptidase-4 (DPP4) inhibitor sitagliptin reduced blood glucose levels, increased GLP1 levels and prevented T2DM-induced DiaCM in mice by shifting the energy substrate utilization in the heart from fatty acids towards glucose [41,42]. Recently, sodium-glucose cotransporter type 2 (SGLT-2) inhibitors, novel hypoglycemic agents that increase urinary Na+ and glucose excretion, were introduced to DM and DiaCM research and have come into the spotlight. In addition to the beneficial effects of SGLT-2 inhibitors on glucose-lowering or natriuretic action, several potential cardioprotective mechanisms of SGLT-2 inhibitors have been reported [5,13,43]. A number of studies have shown the multiple effects of SGLT-2 inhibitors on cardiac iron homeostasis, antioxidative stress, anti-inflammation, RAAS activity, antifibrosis, and GlcNAcylation, as well as mitochondrial function in the heart [43-47]. Excessive O-GlcNAcylation following chronic activation of the hexosamine biosynthetic pathway is associated with posttranslational modifications in the diabetic heart. O-GlcNAcylation impairs cardiac mitochondrial function, Ca2+ homeostasis, and ER stress in DM. A previous study showed that dapagliflozin prevented DiaCM by reducing the levels of O-GlcNAcylated protein in diabetic mice. These results demonstrated that O-GlcNAcylated levels of FOXO1 reduced by SGLT-2 inhibitors contributed to attenuation of DiaCM and improvement in heart function [43,46].
- Oxidative stress
- Excess generation of reactive oxygen species (ROS) or reactive nitrogen species (RNS) is considered to be a central mechanism for diabetes-associated inflammation and remodeling in the heart [13,48,49] and contributes to oxidative stress during both the early and late stages of DiaCM [50,51]. Defects in the antioxidant defense system further increase oxidative stress during the later stages of DiaCM [50,51]. Superoxide dismutase (SOD) has an important role in preventing cardiac damage in the setting of DM. Injection of the SOD mimic mitochondria-targeted mitochondrial triphenylphosphonium chloride (mito-TEMPO) prevented the hyperglycemia-induced increase in superoxide generation, reduced myocardial hypertrophy and improved myocardial function in STZ-induced T1DM mice and db/db T2DM mice compared with vehicle treatment [52].
- The transcription factor nuclear factor erythroid 2-related factor 2 (NRF2) is an essential regulator of the antioxidant response with an important role in preventing diabetes-induced oxidative stress and cell death. Isolated cardiomyocytes from Nrf2 knockout (KO) mice were more susceptible to high glucose-induced cell death than wild-type (WT) cells [13,53]. Furthermore, NRF2-deficient mice were more susceptible to diabetes-induced or angiotensin (Ang) II-induced cardiomyopathy than WT mice, whereas cardiomyocyte-specific overexpression of Nrf2 conferred resistance to Ang II-induced cardiomyopathy [54,55]. Naturally occurring activators of NRF2 have been shown to ameliorate diabetes-induced cardiac complications. Sulforaphane is an organosulfur compound derived from cruciferous vegetables such as cabbage, Brussels sprouts, and broccoli that has been shown to upregulate the expression of numerous genes encoding antioxidant proteins by activating NRF2 signaling [13,56]. The cardioprotective benefits of sulforaphane in attenuating fibrosis, oxidative damage, inflammation, hypertrophy, and cardiac dysfunction have been demonstrated in both T1DM and T2DM mouse models and in mice exposed to Ang II [54,55,57,58]. Administration of the antioxidant N-acetylcysteine (NAC) for 5 weeks to rat and mouse models of STZ-induced T1DM normalized the levels of oxidative stress and subsequently prevented the development of DiaCM [59,60]. Interestingly, the earlier the NAC treatment protocol was initiated after induction of diabetes with STZ during the 12-week experiment, the greater the protection against DiaCM [60], suggesting that early damage mediated by increased oxidative stress has a more important role in the development of DiaCM. In diabetic rats, NAC treatment attenuated cardiac dysfunction and damage after myocardial ischemia–reperfusion injury [61,62].
- Myocardial fibrosis and hypertrophy
- Systemic inflammation, hyperglycemia, and dyslipidemia associated with DM lead to the development of cardiac fibrosis and hypertrophy, which increase myocardial stiffness and result in LV diastolic and systolic dysfunction [13].
- In DiaCM, increased collagen accumulation is observed in perivascular loci, intermyofiber spaces, and replacement fibrosis [14]. Thus, cardiac fibrosis increased in some animal models of both T1DM [14,63-66] and T2DM [67,68]. Under diabetic conditions, advanced glycation end products created by the exposure of proteins and lipids to high glucose levels crosslink extracellular matrix (ECM) proteins, impair ECM degradation by matrix metalloproteinases and increase cardiac stiffness, which together manifest as early LV diastolic dysfunction [13,69,70]. Genetically obese mice exhibited severe diastolic dysfunction, as evidenced by decreasing the ratio of the early (E) to late (A) (E/A) velocities in db/db and ob/ob mice [21,71,72]. Contractile properties were still slightly affected in ob/ob mice [75], while db/db mice displayed reduced fractional shortening and velocity of circumferential fiber shortening at 12 weeks of age [21,72].
- Epicardial and endothelial cells can also contribute to the development of cardiac fibrosis through epithelial-to-mesenchymal or endothelial-to-mesenchymal transition to myofibroblasts [13,73-75].
- The antifibrotic agent cinnamoyl anthranilate reduced collagen production stimulated by transforming growth factor β (TGF-β) signaling in cultured renal mesangial cells [76]. Administration of FT23 and FT011, which are derivatives of cinnamoyl anthranilate, attenuated cardiac structural and functional abnormalities in an animal model of DiaCM [77,78].
- Inflammation and cytokines
- In the diabetic heart, chemokines, cytokines, and exosomes secreted by inflammatory cells contribute to the development of cardiomyocyte hypertrophy and ECM remodeling. Several myocardial processes are activated by a number of proinflammatory factors, dyslipidemia, hyperglycemia, and elevated Ang II levels that are upregulated in the setting of DM [13]. Together, these factors promote the infiltration and accumulation of proinflammatory lymphocytes and macrophages into the lesion site. These inflammatory cells secrete cytokines such as TGF-β, interleukin (IL)-1β, tumor necrosis factor (TNF), IL-6, and interferon-γ that can cause or exacerbate myocardial injury, contributing to further adverse cardiac remodeling [79,80].
- Mice with STZ-induced T1DM have higher T cell infiltration into the myocardium, which is associated with increased myocardial fibrosis and LV dysfunction, than control mice [81]. Inhibition of T cell trafficking in diabetic mice prevented myocardial fibrosis and cardiac dysfunction [82,83].
- Toll-like receptor 4 (TLR4) is expressed in cardiomyocytes, inflammatory cells, and cardiac fibroblasts in both normal and failing hearts [13]. The role of TLR4-mediated inflammatory signaling in the development of DiaCM has been reported in animal models of T1DM and T2DM [84,85]. Inflammatory factors, including nuclear factor-κB and TNF, and protein kinases, such as c-Jun N-terminal kinase (JNK) and p38 mitogen-activated protein kinase (MAPK), can directly lead to cardiomyocyte hypertrophy and can advance myocardial fibrosis [86,87]. Activation of the NLR family pyrin domain containing 3 (NLRP3) inflammasome, a regulator of cell death and inflammation [88], has been associated with cardiac inflammation, fibrosis, and cell death triggered by HFD and STZ administration in a rat model of T2DM [89]. These effects were attenuated by microRNA (miRNA)-mediated Nlrp3 silencing [89] or by pharmacological suppression of NLRP3 inflammasome activation [90].
- Suppression of TLR4 signaling with triptolide or matrine improved cardiac LV function and reduced collagen accumulation in rat models of DiaCM [91,92]. Long-term blockade of TLR4 with the TLR4 inhibitor TAK-242 (also known as CLI-095) was associated with a slight improvement in diabetes-induced erectile dysfunction in rats compared with no treatment, mediated by an increase in cyclic guanosine monophosphate levels and the attenuation of oxidative stress in penile tissue [93]. Numerous small-molecule inhibitors of the NLRP3 inflammasome have evolved in the past several years. The orally active NLRP3 inhibitor 16673-34-0 prevented Western diet-induced systolic and diastolic LV dysfunction in obese mice [94].
- Cardiomyocyte damage and apoptosis
- Apoptosis is an extremely controlled mechanism of programmed cell death and seems to be the principal form of cell death in DiaCM, compared with lower rates due to necrosis [95,96].
- In T1DM animals, both increased death receptor signaling and mitochondria-dependent proapoptotic signaling led to elevated apoptosis in DiaCM, and antioxidant treatment diminished both of these signaling pathways and apoptosis, suggesting an essential role of increased ROS in apoptosis induction in DiaCM [95,97]. A recent study also proposed that dissociation of B-cell lymphoma 2 (Bcl-2) protein from beclin-1 by restoration of impaired AMP-dependent protein kinase (AMPK) activity may decrease apoptosis in DiaCM by restoring autophagy, supporting the suggestion that an interplay between apoptosis and autophagy may be important in DiaCM [98]. Furthermore, ER stress may encourage apoptosis in DiaCM by activating JNK signaling and apoptosis via the extrinsic and intrinsic pathways or by increasing protein kinase RNA-like ER kinase (PERK)-C/EBP homologous protein (CHOP) signaling, which may provoke apoptosis by switching expression towards proapoptotic Bcl-2 proteins [99].
- Impaired CA2+ handling
- In DM, the process of cardiac calcium cycling (Ca2+ entry, intracellular Ca2+ concentration, and Ca2+ efflux) is modified in both humans and animal models, contributing to impaired cardiac contraction and relaxation. Decreased Ca2+ entry is the consequence of both altered voltage dependence of the L-type calcium channel (LTCC) and reduced expression [95]. Impaired intracellular Ca2+ cycling consists of reductions in the amplitude of Ca2+ and in the systolic rate of the Ca2+ rise and fall [100,101]. Prolonged rates of Ca2+ decay may arise from impaired sarco/endoplasmic reticulum Ca2+-ATPase 2a (SER-CA2a) activity during the diastolic period, which may cause a decrease in sarcoplasmic reticulum (SR) Ca2+ storage of up to 50% and, thus, can lead to diastolic dysfunction and impaired relaxation [102].
- In models of T2DM, contractile dysfunction may be driven by a significant decrease in the Ca2+ transient due to reduced Ca2+ influx as a consequence of decreased LTCC expression, by decreased SR Ca2+ content due to increased phospholamban expression and decreased SERCA2a expression, and by the diminished activity and content of ryanodine receptor (RyR) [95,103]. In addition, hyperglycemia may lead to the O-GlcNAcylation of Ca2+/calmodulin-dependent protein kinase II (CaMKII), which may accelerate diastolic SR Ca2+ leakage via RyRs, leading to SR Ca2+ depletion [104].
- A potent late Na+ current inhibitor, ranolazine, might normalize altered intracellular Ca2+ levels in cardiomyocytes due to the close relationship between Ca2+ and Na+ coupling handled by the Na+/Ca2+ exchanger [5,105]. Ranolazine improved several hemodynamic parameters but not cardiac relaxation variables. This result showed that a single treatment using ranolazine is probably not sufficient to influence myocardial structure and cardiac function [5,105].
- Renin-angiotensin-aldosterone system activation
- Current evidence from animal experiments and human patients has identified a critical role for RAAS in DiaCM [5]. Cytoplasmic Ang II enhances cell growth in animal models. Ang II has a definite influence on cell signaling, resulting in cardiomyocyte hypertrophy and proliferation of cardiac fibroblasts [106]. Other factors, such as inflammation, oxidative stress, and aldosterone, may potentiate the harmful effects of Ang II on the heart that lead to myocardial damage in DM [107]. Moreover, the enhanced activation of Ang II and mineralocorticoid receptor signaling might promote insulin resistance by initiating the mammalian target of rapamycin (mTOR)-S6 kinase 1 signal transduction pathway [5,108].
- Recently, renin inhibitors (aliskiren), angiotensin II receptor blockers (ARBs), and angiotensin converting enzyme inhibitors (ACEis) were shown to be protective medications against DiaCM in rat models [5,109]. ACEis and ARBs were also useful agents in both human and animal models of DiaCM [110,111]. The favorable effect of β-adrenoreceptor blockers was also demonstrated in experimental models of DiaCM [112].
- Mitochondrial dysfunction
- Mitochondrial dysfunction is a well-known feature of DiaCM in both animal and human DM. Mitochondrial dysfunction refers to abnormal mitochondrial ultrastructure, increased mitochondrial oxidative stress, impaired activity of Ca2+-sensitive dehydrogenases and F0F1-ATPase, increased sensitivity for Ca2+-induced opening of the mitochondrial permeability transition pore, transcriptional and translational downregulation of oxidative phosphorylation (OXPHOS) subunits, and impaired mitochondrial respiratory capacity and coupling [95,113].
- In humans, several studies have demonstrated mitochondrial dysfunction in the atrium and atrial appendages of DM patients [114-116], with impaired respiration rates and electron transport chain complex activities in patients with DM. In a diabetic mouse model, as early as 1985, an impairment in state 3 respiration of isolated cardiac mitochondria was observed [117]. Since then, mitochondrial dysfunctions have been reported in numerous diabetic rodent models [118]. In terms of T1DM models, STZ-treated rats showed reduced antioxidant glutathione, increased ROS production, and ultimately loss of mitochondrial membrane potential [119]. OVE26 mice also displayed a reduction in glutathione, altered mitochondrial function, and an increase in mitochondrial biogenesis [120]. Akita mice revealed an increased volume of mitochondria with reduced crista densities and respiratory defects [121]. In T2DM models, db/db mice displayed increases in O2 consumption, lipid peroxidation, and mitochondrial ROS generation [122]. Otsuka Long-Evans Tokushima fatty (OLETF) and ob/ob mouse models maintained unchanged levels of uncoupled proteins despite mitochondrial dysfunction [123,124]. Zucker diabetic fatty (ZDF) rats showed increased lipid peroxidation and mitochondrial ROS production rates with elevated antioxidant levels [125,126]. Goto-Kakizaki (GK) and OLETF rats also revealed higher lipid peroxidation and mitochondrial ROS production [125,127,128]. The activity of Sirtuin 3 (SIRT3), a major regulator of intramitochondrial protein acetylation and NAD+-dependent mitochondrial deacetylase, may be reduced in the diabetic heart, causing ROS deposition due to increased acetylation and, thus, suppression of manganese superoxide dismutase (MnSOD) [95,129]. Furthermore, SIRT3 deficiency seems to exacerbate suppression of mitophagy and autophagy in the diabetic heart, whereas SIRT3 overexpression promoted mitophagy and autophagy, attenuated cardiomyocyte apoptosis and diminished mitochondrial defects [130].
- MicroRNAs
- In DiaCM, dysregulations of 316 out of 1,008 total miRNAs were discovered, and pathway analysis demonstrated that several miRNAs are involved in cardiac hypertrophy, oxidative stress, apoptosis, and autophagy [95,131].
- Adenovirus-mediated rescue of the myocardial proviral integration site for Moloney murine leukemia virus-1 (Pim-1) expression in vivo improved systolic and diastolic function, attenuated apoptosis and fibrosis, attenuated ventricular dilation, and restored SERCA2a content in DiaCM [132,133]. The expression of miR-133 is decreased in the STZ-induced diabetic mice, and miR-133 has direct inhibitory effects on collagen deposition by deteriorating connective tissue growth factor expression, indicating that increased miR-133 concentrations may attenuate myocardial fibrosis in DiaCM [134]. Myocardial expression of miR-451 is distinctly increased in mice fed a HFD, and cardiomyocyte-specific deletion of miR-451 decreases ceramide deposition, cardiac hypertrophy, and myocardial fibrosis in this mouse model. Diminution of hypertrophy may come from restoration of attenuated AMPK activity, which may normalize increased mTOR phosphorylation and thus restrict HFD-induced cardiomyocyte growth [135]. Upregulation of miR-30d in DiaCM was suggested to reduce FoxO3a signaling, causing caspase 1 activation and increasing inflammatory signaling, thus resulting in pyroptosis [136]. Based on the various characteristics and mechanisms of DiaCM that can be controlled by miRNAs, a significant contribution of miRNAs to the development of DiaCM was suggested [137].
ANIMAL MODELS ACCORDING TO PATHOMECHANISMS OF DIABETIC CARDIOMYOPATHY
- Clinically, diastolic HF and HFpEF are not synonymous [138,139]. HFpEF is a clinical term used to imply HF with normal systolic function but without any consideration of diastolic function [140,141]. Although diastolic HF and HFpEF are not synonymous, many clinical features overlap [140-142]. Chronic prolonged diastolic dysfunction is a clear risk factor for HFpEF [140,141]. For this reason, an experimental model of human HFpEF generally requires evaluations of ventricular hypertrophy, diastolic dysfunction, exercise intolerance, and interstitial fibrosis [143]. Several established rodent models of HFpEF are summarized in Table 2 [138,144-149].
- A rodent model of HFpEF represents cardiac stiffness and hypertrophy with interstitial fibrosis [143]. Hence, the measurement of LV wall thickness is obligatory to indicate hypertrophy. A number of studies have used LV free wall thickness to demonstrate hypertrophy. Moreover, myocardial interstitial fibrosis and perivascular fibrosis are usually present with hypertrophy [150]. Because a major component of myocardial fibrosis is the presence of collagen in the ECM, collagen staining can manifest its severity [138,151].
- Since DM and obesity are notable comorbidities in HFpEF [152], genetically modified db/db [153] or ob/ob [154] mice are widely applied for cardiometabolic explorations. Diastolic dysfunction of LV has been described for both models [71,155,156]. Additional rodent models for insulin resistance and T2DM include Zucker fatty (ZF) rats, which represent nonfunctional leptin receptors [157], and ZDF rats, which are further inbred strains of ZF rats with high serum glucose concentrations [158]. Recently, Schiattarella et al. [159] created a nongenetic and noninvasive modified model of HFpEF that combined hypertension and hyperlipidemia. They administered a HFD with a nitric oxide synthase inhibitor ad libitum. After 5 weeks, they verified significant impairment of diastolic function, with exercise intolerance and pulmonary congestion. At 15 weeks, significant symptoms and signs of HFpEF had developed [159]. This novel model mimics human pathophysiology, suggesting its appropriateness for use in future research [138].
ANIMAL MODELS FOR HEART FAILURE WITH PRESERVED EJECTION FRACTION
- Innumerable small animal models have been created to explore the impacts of T1DM and T2DM on the heart [14,155]. However, animal models have some limitations and differences from human patients. First, in general, rodents have very similar or identical genetic backgrounds, which is the main limitation, as the models do not reflect human genetic heterogeneity [17,20]. The second limitation of animal models is the rapid induction of stress factors, which is in contrast to the generally slow progression of disease in the human population [17,155]. Third, of several differences between murine and human hearts, heart rate is the fundamental difference between them. On the basis of these contractile kinetics, the ability to increase heart rates in small animal models is impaired compared with humans, which can usually increase by up to nearly threefold. Conversely, the heart rate of mice can increase by approximately 30% to 40% under exercise conditions, which restricts cardiac reserve and is a crucial consideration in the design of animal experiments [155].
- Fourth, the STZ-induced diabetic model can reproduce most characteristics of human DiaCM associated with T1DM. However, STZ may also cause damage to nonpancreatic tissues such as the brain, and the accentuated and rapid onset of T1DM can differ from the disease onset in humans [17,21]. In relation to current data, the most appropriate T1DM model produced by chemical induction of pancreatic toxicity is achieved by STZ. However, the STZ-induced model is not a mutation-induced model. Therefore, it is not an appropriate rodent model to investigate glucose-related gene research, such as glucokinase and unique mutations in glucose-related genes [20,21]. In addition, animal models developing T1DM via point mutations, such as the OVE26 mouse model, also have fundamental limitations in terms of different mechanisms from human T1DM caused by autoimmune failure [21]. The lack of insulin production in the Akita model causes some unusual responses in the heart, such as the absence of fibrosis and hypertrophy and constant levels of oxidation despite mitochondrial dysfunction [20,21]. The GK rat model develops hyperglycemia, insulin resistance, and dyslipidemia with cardiac dysfunction, but obesity and steatosis, commonly observed in clinical practice, are not displayed well [19-21]. Consequently, considering the various limitations mentioned above, an ideal T1DM and T2DM rodent model should be generated in the future.
THE DIFFERENCES BETWEEN HUMAN PATIENTS AND ANIMAL MODELS IN DIABETIC CARDIOMYOPATHY
- Animal models for DM, especially rats and mice, replicate many aspects of the pathogenesis of DiaCM and help to interpret potential contributing mechanisms of the disease [17,155]. A number of experimental protocols have been created to induce DM using supplemented diets, genetics and chemical-induced models [1,17,20]. STZ, a pancreatic β-cell toxin, is commonly used to induce β-cell necrosis and subsequent insulin deficiency [1,10,160]. Although this model does not replicate the clinically more prevalent T2DM, the STZ model evades confounding factors such as obesity and insulin resistance, which need to be taken into consideration in the common genetic models of T2DM, including spontaneous mutation of diabetic db/db and ob/ob mice [1,17]. Recently, there has been a development of T2DM models incorporating low-dose STZ with dietary intervention, as HFD alone is not enough to induce DM [17,20,161]. Remarkably, the use of low-dose STZ and HFD in a rat model mimics late-stage T2DM, when pancreatic β-cell dysfunction becomes obvious [162]. Another model of T1DM is the OVE26 mouse, which overexpresses calmodulin (a multifunctional Ca2+-binding messenger protein) in pancreatic β-cells, resulting in pancreatic β-cell injury [14]. T1DM Akita mice (Ins2Akita+/−) exhibit a spontaneous mutation in the insulin 2 gene, which promotes misfolding of the insulin protein, ER stress, and ultimately β-cell failure [20,163].
- Commonly used composite transgenic models for obesity, insulin resistance, and T2DM are db/db [153] or ob/ob [154] mice, which are based on leptin resistance or deficiency, respectively. ZF rats manifest obesity as a consequence of nonfunctional leptin receptors [164]. ZDF rats were produced by multiple further rounds of inbreeding ZF rats with high serum glucose levels [158]. GK rats are a spontaneous model of T2DM without obesity and are a selective inbred strain derived from Wistar rats [20,165]. Transgenic rodent models that mimic aspects of DiaCM have been created. For example, mice with adipose tissue-specific overexpression of sterol regulatory element-binding protein-1c (SREBP-1c) exhibit elevated plasma triglyceride levels and insulin resistance [14,166]. In addition, mice with cardiomyocyte-specific overexpression of the transcription factor PPARα driven by the α myosin heavy chain gene promoter (MHC-PPARα) display an increase in cardiac fatty acid oxidation and a similar phenotype as DiaCM [167]. Cardiomyocyte-selective insulin receptor KO (CIRKO) mice were used to investigate the effect of decreased insulin signaling in cardiomyocytes without causing systemic metabolic disturbances [168].
- For T2DM-linked DiaCM, since human T2DM usually occurs from unknown polygenic mutations with/without an unhealthy lifestyle, HFD animal models can be closer to ideal T2DM models that develop point mutations in the leptin system or in lipid storage. Polygenetic mutations of obesity, such as OLETF rats and yellow obese gene transgenic Kuo Kondo (KK-Ay) mice, should be further investigated, and nonobese polygenetically mutated GK rats could promote the study of T2DM itself [17,20,21].
- There are several limitations to animal model research; thus, it is necessary to carefully interpret the results from the studies conducted. Fundamentally, genetic, structural, and immune system differences exist between humans and animals; thus, there is a limit to accurately predicting clinical results through diabetic animal models. In particular, it should be considered that different species of animals have different sensitivities to environmental factors, which may result in different symptoms and mechanisms of DiaCM. Despite these specific limitations, animal models, especially rats and mice, serve as inestimable creatures that have greatly developed our understanding of the pathogenesis of DiaCM or HF. Based on recent progress in genome editing, it is highly likely that innumerable novel transgenic models will be created in the near future [155].
- To date, no agreement has been reached on the appropriate management strategy to prevent or treat cardiovascular complications associated with DM. The current treatment regimens for patients with T1DM or T2DM aim to treat insulin resistance, lower inflammation and reduce oxidative stress, which all contribute to the pathogenesis of DiaCM [13]. We hope that the development of an optimal animal model will aid in increasing the understanding of some pathophysiological mechanisms based on the accelerated progression of diabetic complications. Moreover, these models will continue to facilitate the discovery of novel targets and to advance unconventional treatment strategies for HF with DM patients, such as gene- or cell-based therapies [1,5,13,155].
SUMMARY AND PERSPECTIVES
-
CONFLICTS OF INTEREST
No potential conflict of interest relevant to this article was reported.
-
FUNDING
This study was supported by grants from the Basic Science Research Program through the National Research Foundation of Korea (2020R1A2C1009647 to Wang-Soo Lee and 2020R1A2C1010217 to Jaetaek Kim).
NOTES
-
Acknowledgements
- None
| Model | Species | Intervention | Manipulation | Target | DM onset | Phenotypes | |
|---|---|---|---|---|---|---|---|
| Type 1 DM | |||||||
| STZ [14] | Mice | Pharmacological | Injection | β-Cell | 2 day | Necrosis & loss of insulin production, hyperlipidemia | |
| Alloxan [20] | Mice | Pharmacological | Injection | β-Cell | 5 day | Necrosis & loss of insulin production, high TG | |
| OVE26 [21] | Mice | Transgenic | Overexpression | Calmodulin | 2–3 wk | β-cell damage, high TG | |
| NOD [17] | Mice | Transgenic | Insulitis | β-Cell | 30 wk | β-cell failure, high TG | |
| Akita [14,17] | Mice | Transgenic | Spontaneous missense mutation | Insulin-2 gene | 5–6 wk | Misfolding of insulin protein, facilitate ER stress, β-cell failure, high TG | |
| Type 2 DM | |||||||
| HFD/HSD [14,21] | Mice | Diet-induced | Feeding | 1 wk | Obesity, high TG | ||
| HFD+low dose STZ [14,21] | Mice | Diet & Pharmacological | Feeding+injection | β-Cell | 2–10 wk | Obesity, IR | |
| ob/ob [14,21] | Mice | Transgenic | Deficiency | Leptin | 8–15 wk | Obesity, IR, high TG, FFA | |
| db/db [14,21] | Mice | Transgenic | Nonfunctioning | Leptin receptor | 4–8 wk | Obesity, IR, high TG, FFA | |
| ZF/ZDF [14,21] | Rats | Transgenic | Nonfunctioning | Leptin receptor | 14 wk | Obesity, high TG | |
| GK [18,20] | Rats | Transgenic | Overexpression | SREBP-1c | 3 wk | IR, high TG, FFA | |
| OLETF [20,21] | Rats | Polygenic | Food-intake control defect | CCK-1R, Odb2 | 18 wk | Obesity, high TG | |
| KK-Ay [17,20] | Mice | Polygenic | Spontaneous | Agouti gene | 8–16 wk | Obesity, high TG, IR | |
| NZO/HiLt (male) [17,18] | Mice | Polygenic | Spontaneous | Ab to leptin transporter | 12–24 wk | Obesity, leptin resistance, IR | |
| TallyHo/JngJ (male) [17,18] | Mice | Polygenic | Spontaneous | Tanidd1-3 | 10–16 wk | Obesity, hyperlipidemia, hyperinsulinemia | |
| NONcNZO10/LtJ [18,20] | Mice | Polygenic | Spontaneous | Zinc homeostasis or glucose metabolism | 8–24 wk | Obesity, IR | |
DM, diabetes mellitus; STZ, streptozotocin-induced mice; TG, triglyceride; OVE26, OVE26 diabetic mice; NOD, nonobese diabetic mice; Akita, a C57BL/6NSlc mouse with a spontaneous mutation in the insulin‑2 gene; ER, endoplasmic reticulum; HFD/HSD, high-fat/high-sucrose diet; IR, insulin resistance; ob/ob, leptin-deficient mice; FFA, free fatty acid; db/db, leptin receptor-deficient mice; ZF, Zucker fatty rats; ZDF, Zucker diabetic fatty rats; GK, Goto-Kakizaki rats; CCK-1R, cholecystokinnin-1 receptor; Odb2, diabetogenic gene located on chromosome 14; SREBP-1c, sterol regulatory element-binding protein-1c; OLETF, Otsuka Long-Evans Tokushima fatty rats; KK-Ay, yellow obese gene transgenic Kuo Kondo mice; NZO, New Zealand obese mice; Ab, antibody; Tanidd1, a mouse chromosome 19 quantitative trait loci associated with diabetes in TALLYHO mice; NONcNZO10/LtJ, a recombinant congenic strain comprising approximately 88% genome contribution from the NON/LtJ (nonobese and nondiabetic) strain and 12% from the New Zealand obese strain.
| Strain | Model | Manipulation | Phenotypes | |
|---|---|---|---|---|
| Mice | ||||
| ob/ob | Obesity [138,153] | Hypertrophy, diastolic dysfunction | ||
| db/db | DM [138,154] | IR, hypertrophy, diastolic dysfunction | ||
| SAMP8 | Aging [144] | Diastolic dysfunction | ||
| C57BL/6 | Obesity+HTN [145] | HFD+L-NAME | Hypertrophy, diastolic dysfunction, pulmonary congestion | |
| HTN [146] | TAC | Hypertrophy, fibrosis, diastolic dysfunction | ||
| HTN [138,147] | Aldosterone, unilateral nephrectomy, 1% NaCl water-drinking | Hypertrophy, fibrosis, diastolic dysfunction | ||
| HTN [145,148] | DOCA salt, unilateral nephrectomy, 1% NaCl water-drinking | Mild HTN, hypertrophy | ||
| Rats | ||||
| Wistar | HTN [149] | DOCA salt, unilateral nephrectomy, 1% NaCl water-drinking | Severe HTN, hypertrophy | |
| DSS | HTN [138,148] | 4%–8% NaCl chow | Severe HTN, diastolic HF | |
ob/ob, mice with leptin deficiency; db/db, mice with leptin receptor deficiency; DM, diabetes mellitus; IR, insulin resistance; SAMP8, mice with senescence-accelerated mouse prone 8; HTN, hypertension; HFD, high-fat diet; L-NAME, L-NG-nitroarginine methyl ester; TAC transverse aortic constriction; DOCA, deoxycorticosterone acetate; DSS, Dahl salt-sensitive rats; HF, heart failure.
- 1. Tate M, Prakoso D, Willis AM, Peng C, Deo M, Qin CX, et al. Characterising an alternative murine model of diabetic cardiomyopathy. Front Physiol 2019;10:1395.ArticlePubMedPMC
- 2. Ogurtsova K, da Rocha Fernandes JD, Huang Y, Linnenkamp U, Guariguata L, Cho NH, et al. IDF diabetes atlas: global estimates for the prevalence of diabetes for 2015 and 2040. Diabetes Res Clin Pract 2017;128:40-50.ArticlePubMed
- 3. Retnakaran R, Cull CA, Thorne KI, Adler AI, Holman RR; UKPDS Study Group. Risk factors for renal dysfunction in type 2 diabetes: U.K. prospective diabetes study 74. Diabetes 2006;55:1832-9.PubMed
- 4. Semeraro F, Cancarini A, dell’Omo R, Rezzola S, Romano MR, Costagliola C. Diabetic retinopathy: vascular and inflammatory disease. J Diabetes Res 2015;2015:582060.ArticlePubMedPMCPDF
- 5. Lee WS, Kim J. Diabetic cardiomyopathy: where we are and where we are going. Korean J Intern Med 2017;32:404-21.ArticlePubMedPMCPDF
- 6. Maisch B, Alter P, Pankuweit S. Diabetic cardiomyopathy: fact or fiction? Herz 2011;36:102-15.ArticlePubMedPDF
- 7. Lourenco AP, Leite-Moreira AF, Balligand JL, Bauersachs J, Dawson D, de Boer RA, et al. An integrative translational approach to study heart failure with preserved ejection fraction: a position paper from the Working Group on Myocardial Function of the European Society of Cardiology. Eur J Heart Fail 2018;20:216-27.ArticlePubMedPDF
- 8. Seferovic PM, Petrie MC, Filippatos GS, Anker SD, Rosano G, Bauersachs J, et al. Type 2 diabetes mellitus and heart failure: a position statement from the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail 2018;20:853-72.ArticlePubMedPDF
- 9. Russo I, Frangogiannis NG. Diabetes-associated cardiac fibrosis: cellular effectors, molecular mechanisms and therapeutic opportunities. J Mol Cell Cardiol 2016;90:84-93.ArticlePubMed
- 10. Tate M, Grieve DJ, Ritchie RH. Are targeted therapies for diabetic cardiomyopathy on the horizon? Clin Sci (Lond) 2017;131:897-915.ArticlePubMedPDF
- 11. Pappachan JM, Varughese GI, Sriraman R, Arunagirinathan G. Diabetic cardiomyopathy: pathophysiology, diagnostic evaluation and management. World J Diabetes 2013;4:177-89.ArticlePubMedPMC
- 12. Jia G, DeMarco VG, Sowers JR. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat Rev Endocrinol 2016;12:144-53.ArticlePubMedPDF
- 13. Tan Y, Zhang Z, Zheng C, Wintergerst KA, Keller BB, Cai L. Mechanisms of diabetic cardiomyopathy and potential therapeutic strategies: preclinical and clinical evidence. Nat Rev Cardiol 2020;17:585-607.ArticlePubMedPMCPDF
- 14. Riehle C, Bauersachs J. Of mice and men: models and mechanisms of diabetic cardiomyopathy. Basic Res Cardiol 2018;114:2.ArticlePubMedPMCPDF
- 15. Gibbs RA, Weinstock GM, Metzker ML, Muzny DM, Sodergren EJ, Scherer S, et al. Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature 2004;428:493-521.PubMed
- 16. Mouse Genome Sequencing Consortium, Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, et al. Initial sequencing and comparative analysis of the mouse genome. Nature 2002;420:520-62.ArticlePubMedPDF
- 17. Clee SM, Attie AD. The genetic landscape of type 2 diabetes in mice. Endocr Rev 2007;28:48-83.ArticlePubMed
- 18. Joost HG, Schurmann A. The genetic basis of obesity-associated type 2 diabetes (diabesity) in polygenic mouse models. Mamm Genome 2014;25:401-12.ArticlePubMedPMCPDF
- 19. Fang JY, Lin CH, Huang TH, Chuang SY. In vivo rodent models of type 2 diabetes and their usefulness for evaluating flavonoid bioactivity. Nutrients 2019;11:530.ArticlePubMedPMC
- 20. Dhuria RS, Singh G, Kaur A, Kaur R, Kaur T. Current status and patent prospective of animal models in diabetic research. Adv Biomed Res 2015;4:117.ArticlePubMedPMC
- 21. Fuentes-Antras J, Picatoste B, Gomez-Hernandez A, Egido J, Tunon J, Lorenzo O. Updating experimental models of diabetic cardiomyopathy. J Diabetes Res 2015;2015:656795.PubMedPMC
- 22. Cook SA, Varela-Carver A, Mongillo M, Kleinert C, Khan MT, Leccisotti L, et al. Abnormal myocardial insulin signalling in type 2 diabetes and left-ventricular dysfunction. Eur Heart J 2010;31:100-11.ArticlePubMed
- 23. Wright JJ, Kim J, Buchanan J, Boudina S, Sena S, Bakirtzi K, et al. Mechanisms for increased myocardial fatty acid utilization following short-term high-fat feeding. Cardiovasc Res 2009;82:351-60.ArticlePubMedPMC
- 24. Battiprolu PK, Hojayev B, Jiang N, Wang ZV, Luo X, Iglewski M, et al. Metabolic stress-induced activation of FoxO1 triggers diabetic cardiomyopathy in mice. J Clin Invest 2012;122:1109-18.ArticlePubMedPMC
- 25. Wende AR, Kim J, Holland WL, Wayment BE, O’Neill BT, Tuinei J, et al. Glucose transporter 4-deficient hearts develop maladaptive hypertrophy in response to physiological or pathological stresses. Am J Physiol Heart Circ Physiol 2017;313:H1098-108.ArticlePubMedPMC
- 26. Luiken JJ, Koonen DP, Willems J, Zorzano A, Becker C, Fischer Y, et al. Insulin stimulates long-chain fatty acid utilization by rat cardiac myocytes through cellular redistribution of FAT/CD36. Diabetes 2002;51:3113-9.ArticlePubMedPDF
- 27. Luiken JJ, Coort SL, Willems J, Coumans WA, Bonen A, van der Vusse GJ, et al. Contraction-induced fatty acid translocase/CD36 translocation in rat cardiac myocytes is mediated through AMP-activated protein kinase signaling. Diabetes 2003;52:1627-34.ArticlePubMedPDF
- 28. Glatz JF, Luiken JJ. Dynamic role of the transmembrane glycoprotein CD36 (SR-B2) in cellular fatty acid uptake and utilization. J Lipid Res 2018;59:1084-93.ArticlePubMedPMC
- 29. Ouwens DM, Diamant M, Fodor M, Habets DD, Pelsers MM, El Hasnaoui M, et al. Cardiac contractile dysfunction in insulin-resistant rats fed a high-fat diet is associated with elevated CD36-mediated fatty acid uptake and esterification. Diabetologia 2007;50:1938-48.ArticlePubMedPMCPDF
- 30. Lee TW, Bai KJ, Lee TI, Chao TF, Kao YH, Chen YJ. PPARs modulate cardiac metabolism and mitochondrial function in diabetes. J Biomed Sci 2017;24:5.ArticlePubMedPMCPDF
- 31. Du K, Herzig S, Kulkarni RN, Montminy M. TRB3: a tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science 2003;300:1574-7.ArticlePubMed
- 32. Koh HJ, Toyoda T, Didesch MM, Lee MY, Sleeman MW, Kulkarni RN, et al. Tribbles 3 mediates endoplasmic reticulum stress-induced insulin resistance in skeletal muscle. Nat Commun 2013;4:1871.ArticlePubMedPMCPDF
- 33. Gu J, Yan X, Dai X, Wang Y, Lin Q, Xiao J, et al. Metallothionein preserves Akt2 activity and cardiac function via inhibiting TRB3 in diabetic hearts. Diabetes 2018;67:507-17.ArticlePubMedPDF
- 34. Ti Y, Xie GL, Wang ZH, Bi XL, Ding WY, Wang J, et al. TRB3 gene silencing alleviates diabetic cardiomyopathy in a type 2 diabetic rat model. Diabetes 2011;60:2963-74.ArticlePubMedPMCPDF
- 35. Mansor LS, Sousa Fialho MD, Yea G, Coumans WA, West JA, Kerr M, et al. Inhibition of sarcolemmal FAT/CD36 by sulfo-N-succinimidyl oleate rapidly corrects metabolism and restores function in the diabetic heart following hypoxia/reoxygenation. Cardiovasc Res 2017;113:737-48.ArticlePubMedPMC
- 36. Kim SK, Zhao ZS, Lee YJ, Lee KE, Kang SM, Choi D, et al. Leftventricular diastolic dysfunction may be prevented by chronic treatment with PPAR-alpha or -gamma agonists in a type 2 diabetic animal model. Diabetes Metab Res Rev 2003;19:487-93.ArticlePubMed
- 37. Forcheron F, Basset A, Abdallah P, Del Carmine P, Gadot N, Beylot M. Diabetic cardiomyopathy: effects of fenofibrate and metformin in an experimental model: the Zucker diabetic rat. Cardiovasc Diabetol 2009;8:16.ArticlePubMedPMCPDF
- 38. Baraka A, AbdelGawad H. Targeting apoptosis in the heart of streptozotocin-induced diabetic rats. J Cardiovasc Pharmacol Ther 2010;15:175-81.ArticlePubMedPDF
- 39. Liu J, Liu Y, Chen L, Wang Y, Li J. Glucagon-like peptide-1 analog liraglutide protects against diabetic cardiomyopathy by the inhibition of the endoplasmic reticulum stress pathway. J Diabetes Res 2013;2013:630537.ArticlePubMedPMCPDF
- 40. Wu L, Wang K, Wang W, Wen Z, Wang P, Liu L, et al. Glucagon-like peptide-1 ameliorates cardiac lipotoxicity in diabetic cardiomyopathy via the PPARα pathway. Aging Cell 2018;17:e12763.PubMedPMC
- 41. Ramirez E, Picatoste B, Gonzalez-Bris A, Oteo M, Cruz F, Caro-Vadillo A, et al. Sitagliptin improved glucose assimilation in detriment of fatty-acid utilization in experimental type-II diabetes: role of GLP-1 isoforms in Glut4 receptor trafficking. Cardiovasc Diabetol 2018;17:12.ArticlePubMedPMCPDF
- 42. Hamdani N, Hervent AS, Vandekerckhove L, Matheeussen V, Demolder M, Baerts L, et al. Left ventricular diastolic dysfunction and myocardial stiffness in diabetic mice is attenuated by inhibition of dipeptidyl peptidase 4. Cardiovasc Res 2014;104:423-31.ArticlePubMed
- 43. Li N, Zhou H. SGLT2 inhibitors: a novel player in the treatment and prevention of diabetic cardiomyopathy. Drug Des Devel Ther 2020;14:4775-88.PubMedPMC
- 44. Hamouda NN, Sydorenko V, Qureshi MA, Alkaabi JM, Oz M, Howarth FC. Dapagliflozin reduces the amplitude of shortening and Ca(2+) transient in ventricular myocytes from streptozotocin-induced diabetic rats. Mol Cell Biochem 2015;400:57-68.ArticlePubMedPDF
- 45. Lee TM, Chang NC, Lin SZ. Dapagliflozin, a selective SGLT2 inhibitor, attenuated cardiac fibrosis by regulating the macrophage polarization via STAT3 signaling in infarcted rat hearts. Free Radic Biol Med 2017;104:298-310.ArticlePubMed
- 46. Joubert M, Jagu B, Montaigne D, Marechal X, Tesse A, Ayer A, et al. The sodium-glucose cotransporter 2 inhibitor dapagliflozin prevents cardiomyopathy in a diabetic lipodystrophic mouse model. Diabetes 2017;66:1030-40.ArticlePubMedPDF
- 47. Durak A, Olgar Y, Degirmenci S, Akkus E, Tuncay E, Turan B. A SGLT2 inhibitor dapagliflozin suppresses prolonged ventricular-repolarization through augmentation of mitochondrial function in insulin-resistant metabolic syndrome rats. Cardiovasc Diabetol 2018;17:144.ArticlePubMedPMCPDF
- 48. Cai L, Kang YJ. Oxidative stress and diabetic cardiomyopathy: a brief review. Cardiovasc Toxicol 2001;1:181-93.ArticlePubMed
- 49. Wilson AJ, Gill EK, Abudalo RA, Edgar KS, Watson CJ, Grieve DJ. Reactive oxygen species signalling in the diabetic heart: emerging prospect for therapeutic targeting. Heart 2018;104:293-9.ArticlePubMed
- 50. Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000;404:787-90.ArticlePubMedPDF
- 51. Tan Y, Ichikawa T, Li J, Si Q, Yang H, Chen X, et al. Diabetic downregulation of Nrf2 activity via ERK contributes to oxidative stress-induced insulin resistance in cardiac cells in vitro and in vivo. Diabetes 2011;60:625-33.ArticlePubMedPMCPDF
- 52. Ni R, Cao T, Xiong S, Ma J, Fan GC, Lacefield JC, et al. Therapeutic inhibition of mitochondrial reactive oxygen species with mito-TEMPO reduces diabetic cardiomyopathy. Free Radic Biol Med 2016;90:12-23.ArticlePubMed
- 53. He X, Kan H, Cai L, Ma Q. Nrf2 is critical in defense against high glucose-induced oxidative damage in cardiomyocytes. J Mol Cell Cardiol 2009;46:47-58.ArticlePubMed
- 54. Xin Y, Bai Y, Jiang X, Zhou S, Wang Y, Wintergerst KA, et al. Sulforaphane prevents angiotensin II-induced cardiomyopathy by activation of Nrf2 via stimulating the Akt/GSK-3ß/Fyn pathway. Redox Biol 2018;15:405-17.ArticlePubMedPMC
- 55. Gu J, Cheng Y, Wu H, Kong L, Wang S, Xu Z, et al. Metallothionein is downstream of Nrf2 and partially mediates sulforaphane prevention of diabetic cardiomyopathy. Diabetes 2017;66:529-42.ArticlePubMedPDF
- 56. Fahey JW, Talalay P. Antioxidant functions of sulforaphane: a potent inducer of phase II detoxication enzymes. Food Chem Toxicol 1999;37:973-9.ArticlePubMed
- 57. Bai Y, Cui W, Xin Y, Miao X, Barati MT, Zhang C, et al. Prevention by sulforaphane of diabetic cardiomyopathy is associated with up-regulation of Nrf2 expression and transcription activation. J Mol Cell Cardiol 2013;57:82-95.ArticlePubMed
- 58. Zhang Z, Wang S, Zhou S, Yan X, Wang Y, Chen J, et al. Sulforaphane prevents the development of cardiomyopathy in type 2 diabetic mice probably by reversing oxidative stress-induced inhibition of LKB1/AMPK pathway. J Mol Cell Cardiol 2014;77:42-52.ArticlePubMed
- 59. Xia Z, Kuo KH, Nagareddy PR, Wang F, Guo Z, Guo T, et al. N-acetylcysteine attenuates PKCbeta2 overexpression and myocardial hypertrophy in streptozotocin-induced diabetic rats. Cardiovasc Res 2007;73:770-82.ArticlePubMed
- 60. Liu C, Lu XZ, Shen MZ, Xing CY, Ma J, Duan YY, et al. N-acetyl cysteine improves the diabetic cardiac function: possible role of fibrosis inhibition. BMC Cardiovasc Disord 2015;15:84.ArticlePubMedPMCPDF
- 61. Okazaki T, Otani H, Shimazu T, Yoshioka K, Fujita M, Iwasaka T. Ascorbic acid and N-acetyl cysteine prevent uncoupling of nitric oxide synthase and increase tolerance to ischemia/reperfusion injury in diabetic rat heart. Free Radic Res 2011;45:1173-83.ArticlePubMed
- 62. Su W, Zhang Y, Zhang Q, Xu J, Zhan L, Zhu Q, et al. N-acetylcysteine attenuates myocardial dysfunction and postischemic injury by restoring caveolin-3/eNOS signaling in diabetic rats. Cardiovasc Diabetol 2016;15:146.ArticlePubMedPMCPDF
- 63. Candido R, Forbes JM, Thomas MC, Thallas V, Dean RG, Burns WC, et al. A breaker of advanced glycation end products attenuates diabetes-induced myocardial structural changes. Circ Res 2003;92:785-92.ArticlePubMed
- 64. Singh VP, Le B, Khode R, Baker KM, Kumar R. Intracellular angiotensin II production in diabetic rats is correlated with cardiomyocyte apoptosis, oxidative stress, and cardiac fibrosis. Diabetes 2008;57:3297-306.ArticlePubMedPMCPDF
- 65. Van Linthout S, Seeland U, Riad A, Eckhardt O, Hohl M, Dhayat N, et al. Reduced MMP-2 activity contributes to cardiac fibrosis in experimental diabetic cardiomyopathy. Basic Res Cardiol 2008;103:319-27.ArticlePubMedPDF
- 66. Wang Y, Sun W, Du B, Miao X, Bai Y, Xin Y, et al. Therapeutic effect of MG-132 on diabetic cardiomyopathy is associated with its suppression of proteasomal activities: roles of Nrf2 and NF-κB. Am J Physiol Heart Circ Physiol 2013;304:H567-78.ArticlePubMed
- 67. Mizushige K, Yao L, Noma T, Kiyomoto H, Yu Y, Hosomi N, et al. Alteration in left ventricular diastolic filling and accumulation of myocardial collagen at insulin-resistant prediabetic stage of a type II diabetic rat model. Circulation 2000;101:899-907.ArticlePubMed
- 68. Zhou YT, Grayburn P, Karim A, Shimabukuro M, Higa M, Baetens D, et al. Lipotoxic heart disease in obese rats: implications for human obesity. Proc Natl Acad Sci U S A 2000;97:1784-9.ArticlePubMedPMC
- 69. Wan A, Rodrigues B. Endothelial cell-cardiomyocyte crosstalk in diabetic cardiomyopathy. Cardiovasc Res 2016;111:172-83.ArticlePubMedPMC
- 70. Choi SY, Chang HJ, Choi SI, Kim KI, Cho YS, Youn TJ, et al. Long-term exercise training attenuates age-related diastolic dysfunction: association of myocardial collagen cross-linking. J Korean Med Sci 2009;24:32-9.ArticlePubMedPMC
- 71. Christoffersen C, Bollano E, Lindegaard ML, Bartels ED, Goetze JP, Andersen CB, et al. Cardiac lipid accumulation associated with diastolic dysfunction in obese mice. Endocrinology 2003;144:3483-90.ArticlePubMedPDF
- 72. Semeniuk LM, Kryski AJ, Severson DL. Echocardiographic assessment of cardiac function in diabetic db/db and transgenic db/db-hGLUT4 mice. Am J Physiol Heart Circ Physiol 2002;283:H976-82.ArticlePubMed
- 73. Travers JG, Kamal FA, Robbins J, Yutzey KE, Blaxall BC. Cardiac fibrosis: the fibroblast awakens. Circ Res 2016;118:1021-40.ArticlePubMedPMC
- 74. Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med 2007;13:952-61.ArticlePubMedPDF
- 75. Smith CL, Baek ST, Sung CY, Tallquist MD. Epicardial-derived cell epithelial-to-mesenchymal transition and fate specification require PDGF receptor signaling. Circ Res 2011;108:e15-26.ArticlePubMedPMC
- 76. Zammit SC, Cox AJ, Gow RM, Zhang Y, Gilbert RE, Krum H, et al. Evaluation and optimization of antifibrotic activity of cinnamoyl anthranilates. Bioorg Med Chem Lett 2009;19:7003-6.ArticlePubMed
- 77. Zhang Y, Edgley AJ, Cox AJ, Powell AK, Wang B, Kompa AR, et al. FT011, a new anti-fibrotic drug, attenuates fibrosis and chronic heart failure in experimental diabetic cardiomyopathy. Eur J Heart Fail 2012;14:549-62.ArticlePubMedPDF
- 78. Tan SM, Zhang Y, Wang B, Tan CY, Zammit SC, Williams SJ, et al. FT23, an orally active antifibrotic compound, attenuates structural and functional abnormalities in an experimental model of diabetic cardiomyopathy. Clin Exp Pharmacol Physiol 2012;39:650-6.ArticlePubMed
- 79. Biernacka A, Cavalera M, Wang J, Russo I, Shinde A, Kong P, et al. Smad3 signaling promotes fibrosis while preserving cardiac and aortic geometry in obese diabetic mice. Circ Heart Fail 2015;8:788-98.ArticlePubMedPMC
- 80. Bajpai A, Tilley DG. The role of leukocytes in diabetic cardiomyopathy. Front Physiol 2018;9:1547.ArticlePubMedPMC
- 81. Lin Y, Tang Y, Wang F. The protective effect of HIF-1α in T lymphocytes on cardiac damage in diabetic mice. Ann Clin Lab Sci 2016;46:32-43.PubMed
- 82. Laroumanie F, Douin-Echinard V, Pozzo J, Lairez O, Tortosa F, Vinel C, et al. CD4+ T cells promote the transition from hypertrophy to heart failure during chronic pressure overload. Circulation 2014;129:2111-24.ArticlePubMed
- 83. Nevers T, Salvador AM, Grodecki-Pena A, Knapp A, Velazquez F, Aronovitz M, et al. Left ventricular T-cell recruitment contributes to the pathogenesis of heart failure. Circ Heart Fail 2015;8:776-87.ArticlePubMedPMC
- 84. Dong B, Qi D, Yang L, Huang Y, Xiao X, Tai N, et al. TLR4 regulates cardiac lipid accumulation and diabetic heart disease in the nonobese diabetic mouse model of type 1 diabetes. Am J Physiol Heart Circ Physiol 2012;303:H732-42.ArticlePubMedPMC
- 85. Tao A, Song J, Lan T, Xu X, Kvietys P, Kao R, et al. Cardiomyocyte-fibroblast interaction contributes to diabetic cardiomyopathy in mice: role of HMGB1/TLR4/IL-33 axis. Biochim Biophys Acta 2015;1852(10 Pt A):2075-85.ArticlePubMed
- 86. Nakamura M, Sadoshima J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat Rev Cardiol 2018;15:387-407.ArticlePubMedPDF
- 87. Gordon JW, Shaw JA, Kirshenbaum LA. Multiple facets of NFκB in the heart: to be or not to NF-κB. Circ Res 2011;108:1122-32.ArticlePubMed
- 88. He Y, Hara H, Nunez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci 2016;41:1012-21.ArticlePubMedPMC
- 89. Luo B, Li B, Wang W, Liu X, Xia Y, Zhang C, et al. NLRP3 gene silencing ameliorates diabetic cardiomyopathy in a type 2 diabetes rat model. PLoS One 2014;9:e104771.ArticlePubMedPMC
- 90. Luo B, Li B, Wang W, Liu X, Liu X, Xia Y, et al. Rosuvastatin alleviates diabetic cardiomyopathy by inhibiting NLRP3 inflammasome and MAPK pathways in a type 2 diabetes rat model. Cardiovasc Drugs Ther 2014;28:33-43.ArticlePubMedPDF
- 91. Liu ZW, Wang JK, Qiu C, Guan GC, Liu XH, Li SJ, et al. Matrine pretreatment improves cardiac function in rats with diabetic cardiomyopathy via suppressing ROS/TLR-4 signaling pathway. Acta Pharmacol Sin 2015;36:323-33.ArticlePubMedPMCPDF
- 92. Guo X, Xue M, Li CJ, Yang W, Wang SS, Ma ZJ, et al. Protective effects of triptolide on TLR4 mediated autoimmune and inflammatory response induced myocardial fibrosis in diabetic cardiomyopathy. J Ethnopharmacol 2016;193:333-44.ArticlePubMed
- 93. Nunes KP, de Oliveira AA, Szasz T, Biancardi VC, Webb RC. Blockade of toll-like receptor 4 attenuates erectile dysfunction in diabetic rats. J Sex Med 2018;15:1235-45.ArticlePubMedPDF
- 94. Carbone S, Mauro AG, Prestamburgo A, Halquist MS, Narayan P, Potere N, et al. An orally available NLRP3 inflammasome inhibitor prevents Western diet-induced cardiac dysfunction in mice. J Cardiovasc Pharmacol 2018;72:303-7.ArticlePubMedPMC
- 95. Gollmer J, Zirlik A, Bugger H. Established and emerging mechanisms of diabetic cardiomyopathy. J Lipid Atheroscler 2019;8:26-47.ArticlePubMedPMCPDF
- 96. Chowdhry MF, Vohra HA, Galinanes M. Diabetes increases apoptosis and necrosis in both ischemic and nonischemic human myocardium: role of caspases and poly-adenosine diphosphate-ribose polymerase. J Thorac Cardiovasc Surg 2007;134:124-31.ArticlePubMed
- 97. Bojunga J, Nowak D, Mitrou PS, Hoelzer D, Zeuzem S, Chow KU. Antioxidative treatment prevents activation of death-receptor- and mitochondrion-dependent apoptosis in the hearts of diabetic rats. Diabetologia 2004;47:2072-80.ArticlePubMedPDF
- 98. He C, Zhu H, Li H, Zou MH, Xie Z. Dissociation of Bcl-2-Beclin1 complex by activated AMPK enhances cardiac autophagy and protects against cardiomyocyte apoptosis in diabetes. Diabetes 2013;62:1270-81.ArticlePubMedPMCPDF
- 99. Yang L, Zhao D, Ren J, Yang J. Endoplasmic reticulum stress and protein quality control in diabetic cardiomyopathy. Biochim Biophys Acta 2015;1852:209-18.ArticlePubMed
- 100. Shao CH, Rozanski GJ, Patel KP, Bidasee KR. Dyssynchronous (non-uniform) Ca2+ release in myocytes from streptozotocin-induced diabetic rats. J Mol Cell Cardiol 2007;42:234-46.ArticlePubMed
- 101. Kranstuber AL, Del Rio C, Biesiadecki BJ, Hamlin RL, Ottobre J, Gyorke S, et al. Advanced glycation end product cross-link breaker attenuates diabetes-induced cardiac dysfunction by improving sarcoplasmic reticulum calcium handling. Front Physiol 2012;3:292.ArticlePubMedPMC
- 102. Lacombe VA, Viatchenko-Karpinski S, Terentyev D, Sridhar A, Emani S, Bonagura JD, et al. Mechanisms of impaired calcium handling underlying subclinical diastolic dysfunction in diabetes. Am J Physiol Regul Integr Comp Physiol 2007;293:R1787-97.ArticlePubMedPMC
- 103. Pereira L, Ruiz-Hurtado G, Rueda A, Mercadier JJ, Benitah JP, Gomez AM. Calcium signaling in diabetic cardiomyocytes. Cell Calcium 2014;56:372-80.ArticlePubMed
- 104. Erickson JR, Pereira L, Wang L, Han G, Ferguson A, Dao K, et al. Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature 2013;502:372-6.ArticlePubMedPMCPDF
- 105. Maier LS, Layug B, Karwatowska-Prokopczuk E, Belardinelli L, Lee S, Sander J, et al. RAnoLazIne for the treatment of diastolic heart failure in patients with preserved ejection fraction: the RALI-DHF proof-of-concept study. JACC Heart Fail 2013;1:115-22.PubMed
- 106. Kumar R, Yong QC, Thomas CM, Baker KM. Intracardiac intracellular angiotensin system in diabetes. Am J Physiol Regul Integr Comp Physiol 2012;302:R510-7.PubMed
- 107. Kurdi M, Booz GW. New take on the role of angiotensin II in cardiac hypertrophy and fibrosis. Hypertension 2011;57:1034-8.ArticlePubMedPMC
- 108. DeMarco VG, Aroor AR, Sowers JR. The pathophysiology of hypertension in patients with obesity. Nat Rev Endocrinol 2014;10:364-76.ArticlePubMedPMCPDF
- 109. Thomas CM, Yong QC, Seqqat R, Chandel N, Feldman DL, Baker KM, et al. Direct renin inhibition prevents cardiac dysfunction in a diabetic mouse model: comparison with an angiotensin receptor antagonist and angiotensin-converting enzyme inhibitor. Clin Sci (Lond) 2013;124:529-41.ArticlePubMedPMCPDF
- 110. Machackova J, Liu X, Lukas A, Dhalla NS. Renin-angiotensin blockade attenuates cardiac myofibrillar remodelling in chronic diabetes. Mol Cell Biochem 2004;261:271-8.ArticlePubMed
- 111. Symeonides P, Koulouris S, Vratsista E, Triantafyllou K, Ioannidis G, Thalassinos N, et al. Both ramipril and telmisartan reverse indices of early diabetic cardiomyopathy: a comparative study. Eur J Echocardiogr 2007;8:480-6.ArticlePubMed
- 112. Sharma V, McNeill JH. Parallel effects of β-adrenoceptor blockade on cardiac function and fatty acid oxidation in the diabetic heart: confronting the maze. World J Cardiol 2011;3:281-302.ArticlePubMedPMC
- 113. Song M, Gong G, Burelle Y, Gustafsson AB, Kitsis RN, Matkovich SJ, et al. Interdependence of Parkin-mediated mitophagy and mitochondrial fission in adult mouse hearts. Circ Res 2015;117:346-51.ArticlePubMedPMC
- 114. Anderson EJ, Kypson AP, Rodriguez E, Anderson CA, Lehr EJ, Neufer PD. Substrate-specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart. J Am Coll Cardiol 2009;54:1891-8.ArticlePubMedPMC
- 115. Montaigne D, Marechal X, Coisne A, Debry N, Modine T, Fayad G, et al. Myocardial contractile dysfunction is associated with impaired mitochondrial function and dynamics in type 2 diabetic but not in obese patients. Circulation 2014;130:554-64.ArticlePubMed
- 116. Croston TL, Thapa D, Holden AA, Tveter KJ, Lewis SE, Shepherd DL, et al. Functional deficiencies of subsarcolemmal mitochondria in the type 2 diabetic human heart. Am J Physiol Heart Circ Physiol 2014;307:H54-65.ArticlePubMedPMC
- 117. Kuo TH, Giacomelli F, Wiener J. Oxidative metabolism of Polytron versus Nagarse mitochondria in hearts of genetically diabetic mice. Biochim Biophys Acta 1985;806:9-15.ArticlePubMed
- 118. Bugger H, Abel ED. Rodent models of diabetic cardiomyopathy. Dis Model Mech 2009;2:454-66.ArticlePubMedPDF
- 119. Ghosh S, Pulinilkunnil T, Yuen G, Kewalramani G, An D, Qi D, et al. Cardiomyocyte apoptosis induced by short-term diabetes requires mitochondrial GSH depletion. Am J Physiol Heart Circ Physiol 2005;289:H768-76.ArticlePubMed
- 120. Song Y, Du Y, Prabhu SD, Epstein PN. Diabetic cardiomyopathy in OVE26 mice shows mitochondrial ROS production and divergence between in vivo and in vitro contractility. Rev Diabet Stud 2007;4:159-68.ArticlePubMedPMC
- 121. Bugger H, Boudina S, Hu XX, Tuinei J, Zaha VG, Theobald HA, et al. Type 1 diabetic akita mouse hearts are insulin sensitive but manifest structurally abnormal mitochondria that remain coupled despite increased uncoupling protein 3. Diabetes 2008;57:2924-32.ArticlePubMedPMCPDF
- 122. Boudina S, Sena S, Theobald H, Sheng X, Wright JJ, Hu XX, et al. Mitochondrial energetics in the heart in obesity-related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes 2007;56:2457-66.PubMed
- 123. Boudina S, Sena S, O’Neill BT, Tathireddy P, Young ME, Abel ED. Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation 2005;112:2686-95.ArticlePubMed
- 124. Boudina S, Abel ED. Diabetic cardiomyopathy revisited. Circulation 2007;115:3213-23.ArticlePubMed
- 125. Vazquez-Medina JP, Popovich I, Thorwald MA, Viscarra JA, Rodriguez R, Sonanez-Organis JG, et al. Angiotensin receptor-mediated oxidative stress is associated with impaired cardiac redox signaling and mitochondrial function in insulinresistant rats. Am J Physiol Heart Circ Physiol 2013;305:H599-607.ArticlePubMedPMC
- 126. Vincent HK, Powers SK, Dirks AJ, Scarpace PJ. Mechanism for obesity-induced increase in myocardial lipid peroxidation. Int J Obes Relat Metab Disord 2001;25:378-88.ArticlePubMedPDF
- 127. Santos DL, Palmeira CM, Seica R, Dias J, Mesquita J, Moreno AJ, et al. Diabetes and mitochondrial oxidative stress: a study using heart mitochondria from the diabetic Goto-Kakizaki rat. Mol Cell Biochem 2003;246:163-70.ArticlePubMed
- 128. Grijalva J, Hicks S, Zhao X, Medikayala S, Kaminski PM, Wolin MS, et al. Exercise training enhanced myocardial endothelial nitric oxide synthase (eNOS) function in diabetic GotoKakizaki (GK) rats. Cardiovasc Diabetol 2008;7:34.ArticlePubMedPMCPDF
- 129. Vazquez EJ, Berthiaume JM, Kamath V, Achike O, Buchanan E, Montano MM, et al. Mitochondrial complex I defect and increased fatty acid oxidation enhance protein lysine acetylation in the diabetic heart. Cardiovasc Res 2015;107:453-65.ArticlePubMedPMC
- 130. Sultana MR, Bagul PK, Katare PB, Anwar Mohammed S, Padiya R, Banerjee SK. Garlic activates SIRT-3 to prevent cardiac oxidative stress and mitochondrial dysfunction in diabetes. Life Sci 2016;164:42-51.ArticlePubMed
- 131. Chen JF, Murchison EP, Tang R, Callis TE, Tatsuguchi M, Deng Z, et al. Targeted deletion of Dicer in the heart leads to dilated cardiomyopathy and heart failure. Proc Natl Acad Sci U S A 2008;105:2111-6.ArticlePubMedPMC
- 132. Costantino S, Paneni F, Luscher TF, Cosentino F. MicroRNA profiling unveils hyperglycaemic memory in the diabetic heart. Eur Heart J 2016;37:572-6.ArticlePubMed
- 133. Katare R, Caporali A, Zentilin L, Avolio E, Sala-Newby G, Oikawa A, et al. Intravenous gene therapy with PIM-1 via a cardiotropic viral vector halts the progression of diabetic cardiomyopathy through promotion of prosurvival signaling. Circ Res 2011;108:1238-51.ArticlePubMed
- 134. Liu X, Liu S. Role of microRNAs in the pathogenesis of diabetic cardiomyopathy. Biomed Rep 2017;6:140-5.ArticlePubMedPMC
- 135. Zheng D, Ma J, Yu Y, Li M, Ni R, Wang G, et al. Silencing of miR-195 reduces diabetic cardiomyopathy in C57BL/6 mice. Diabetologia 2015;58:1949-58.ArticlePubMedPMCPDF
- 136. Raut SK, Kumar A, Singh GB, Nahar U, Sharma V, Mittal A, et al. miR-30c mediates upregulation of Cdc42 and Pak1 in diabetic cardiomyopathy. Cardiovasc Ther 2015;33:89-97.ArticlePubMed
- 137. Li X, Du N, Zhang Q, Li J, Chen X, Liu X, et al. MicroRNA30d regulates cardiomyocyte pyroptosis by directly targeting foxo3a in diabetic cardiomyopathy. Cell Death Dis 2014;5:e1479.ArticlePubMedPMCPDF
- 138. Yoon S, Eom GH. Heart failure with preserved ejection fraction: present status and future directions. Exp Mol Med 2019;51:1-9.ArticlePDF
- 139. Borlaug BA. The pathophysiology of heart failure with preserved ejection fraction. Nat Rev Cardiol 2014;11:507-15.ArticlePubMedPDF
- 140. Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JG, Coats AJ, et al. 2016 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure: the task force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC): developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail 2016;18:891-975.ArticlePubMedPDF
- 141. Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE Jr, Colvin MM, et al. 2017 ACC/AHA/HFSA focused update of the 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology/American Heart Association task force on clinical practice guidelines and the Heart Failure Society of America. Circulation 2017;136:e137-61.ArticlePubMed
- 142. Shah SJ, Katz DH, Selvaraj S, Burke MA, Yancy CW, Gheorghiade M, et al. Phenomapping for novel classification of heart failure with preserved ejection fraction. Circulation 2015;131:269-79.ArticlePubMed
- 143. Valero-Munoz M, Backman W, Sam F. Murine models of heart failure with preserved ejection fraction: a “fishing expedition”. JACC Basic Transl Sci 2017;2:770-89.PubMedPMC
- 144. Zhang L, Li Q, Wolff LT, Antonio GE, Yeung DK, Zhang A, et al. Changes of brain activity in the aged SAMP mouse. Biogerontology 2007;8:81-8.ArticlePubMedPDF
- 145. Grobe JL, Buehrer BA, Hilzendeger AM, Liu X, Davis DR, Xu D, et al. Angiotensinergic signaling in the brain mediates metabolic effects of deoxycorticosterone (DOCA)-salt in C57 mice. Hypertension 2011;57:600-7.ArticlePubMedPMC
- 146. Dunlay SM, Roger VL, Weston SA, Jiang R, Redfield MM. Longitudinal changes in ejection fraction in heart failure patients with preserved and reduced ejection fraction. Circ Heart Fail 2012;5:720-6.ArticlePubMedPMC
- 147. Tanaka K, Wilson RM, Essick EE, Duffen JL, Scherer PE, Ouchi N, et al. Effects of adiponectin on calcium-handling proteins in heart failure with preserved ejection fraction. Circ Heart Fail 2014;7:976-85.ArticlePubMedPMC
- 148. Mohammed SF, Ohtani T, Korinek J, Lam CS, Larsen K, Simari RD, et al. Mineralocorticoid accelerates transition to heart failure with preserved ejection fraction via “nongenomic effects”. Circulation 2010;122:370-8.ArticlePubMedPMC
- 149. Matsumura Y, Kuro T, Konishi F, Takaoka M, Gariepy CE, Yanagisawa M. Enhanced blood pressure sensitivity to DOCA-salt treatment in endothelin ET(B) receptor-deficient rats. Br J Pharmacol 2000;129:1060-2.ArticlePubMedPMCPDF
- 150. Ho CY, Lopez B, Coelho-Filho OR, Lakdawala NK, Cirino AL, Jarolim P, et al. Myocardial fibrosis as an early manifestation of hypertrophic cardiomyopathy. N Engl J Med 2010;363:552-63.ArticlePubMedPMC
- 151. Hadi AM, Mouchaers KT, Schalij I, Grunberg K, Meijer GA, Vonk-Noordegraaf A, et al. Rapid quantification of myocardial fibrosis: a new macro-based automated analysis. Cell Oncol (Dordr) 2011;34:343-54.ArticlePubMedPMC
- 152. Lavie CJ, Milani RV, Ventura HO. Obesity and cardiovascular disease: risk factor, paradox, and impact of weight loss. J Am Coll Cardiol 2009;53:1925-32.PubMed
- 153. Chen H, Charlat O, Tartaglia LA, Woolf EA, Weng X, Ellis SJ, et al. Evidence that the diabetes gene encodes the leptin receptor: identification of a mutation in the leptin receptor gene in db/db mice. Cell 1996;84:491-5.ArticlePubMed
- 154. Edvell A, Lindstrom P. Initiation of increased pancreatic islet growth in young normoglycemic mice (Umea +/?). Endocrinology 1999;140:778-83.ArticlePubMed
- 155. Riehle C, Bauersachs J. Small animal models of heart failure. Cardiovasc Res 2019;115:1838-49.ArticlePubMedPMCPDF
- 156. Nielsen JM, Kristiansen SB, Norregaard R, Andersen CL, Denner L, Nielsen TT, et al. Blockage of receptor for advanced glycation end products prevents development of cardiac dysfunction in db/db type 2 diabetic mice. Eur J Heart Fail 2009;11:638-47.ArticlePubMedPDF
- 157. Phillips MS, Liu Q, Hammond HA, Dugan V, Hey PJ, Caskey CJ, et al. Leptin receptor missense mutation in the fatty Zucker rat. Nat Genet 1996;13:18-9.ArticlePubMedPDF
- 158. Clark JB, Palmer CJ, Shaw WN. The diabetic Zucker fatty rat. Proc Soc Exp Biol Med 1983;173:68-75.ArticlePubMed
- 159. Schiattarella GG, Altamirano F, Tong D, French KM, Villalobos E, Kim SY, et al. Nitrosative stress drives heart failure with preserved ejection fraction. Nature 2019;568:351-6.ArticlePubMedPMCPDF
- 160. Prakoso D, De Blasio MJ, Qin C, Rosli S, Kiriazis H, Qian H, et al. Phosphoinositide 3-kinase (p110α) gene delivery limits diabetes-induced cardiac NADPH oxidase and cardiomyopathy in a mouse model with established diastolic dysfunction. Clin Sci (Lond) 2017;131:1345-60.ArticlePubMedPDF
- 161. Wanrooy BJ, Kumar KP, Wen SW, Qin CX, Ritchie RH, Wong CH. Distinct contributions of hyperglycemia and high-fat feeding in metabolic syndrome-induced neuroinflammation. J Neuroinflammation 2018;15:293.ArticlePubMedPMCPDF
- 162. Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 2003;52:102-10.PubMed
- 163. Yoshioka M, Kayo T, Ikeda T, Koizumi A. A novel locus, Mody4, distal to D7Mit189 on chromosome 7 determines early-onset NIDDM in nonobese C57BL/6 (Akita) mutant mice. Diabetes 1997;46:887-94.ArticlePubMed
- 164. Takaya K, Ogawa Y, Isse N, Okazaki T, Satoh N, Masuzaki H, et al. Molecular cloning of rat leptin receptor isoform complementary DNAs: identification of a missense mutation in Zucker fatty (fa/fa) rats. Biochem Biophys Res Commun 1996;225:75-83.ArticlePubMed
- 165. Gauguier D, Froguel P, Parent V, Bernard C, Bihoreau MT, Portha B, et al. Chromosomal mapping of genetic loci associated with non-insulin dependent diabetes in the GK rat. Nat Genet 1996;12:38-43.ArticlePubMedPDF
- 166. Shimomura I, Hammer RE, Richardson JA, Ikemoto S, Bashmakov Y, Goldstein JL, et al. Insulin resistance and diabetes mellitus in transgenic mice expressing nuclear SREBP-1c in adipose tissue: model for congenital generalized lipodystrophy. Genes Dev 1998;12:3182-94.ArticlePubMedPMC
- 167. Finck BN, Han X, Courtois M, Aimond F, Nerbonne JM, Kovacs A, et al. A critical role for PPARalpha-mediated lipotoxicity in the pathogenesis of diabetic cardiomyopathy: modulation by dietary fat content. Proc Natl Acad Sci U S A 2003;100:1226-31.PubMedPMC
- 168. Belke DD, Betuing S, Tuttle MJ, Graveleau C, Young ME, Pham M, et al. Insulin signaling coordinately regulates cardiac size, metabolism, and contractile protein isoform expression. J Clin Invest 2002;109:629-39.ArticlePubMedPMC
REFERENCES
Figure & Data
References
Citations
- Chitosan Versus Dapagliflozin in a Diabetic Cardiomyopathy Mouse Model
Georgică Târtea, Aurel Popa-Wagner, Veronica Sfredel, Smaranda Ioana Mitran, Alexandra Oltea Dan, Anca-Maria Țucă, Alexandra Nicoleta Preda, Victor Raicea, Eugen Țieranu, Dragoș Cozma, Radu Vătășescu
International Journal of Molecular Sciences.2024; 25(4): 2118. CrossRef - Mitochondrial energy metabolism in diabetic cardiomyopathy: Physiological adaption, pathogenesis, and therapeutic targets
Wanlin Ye, Kun Han, Maodi Xie, Sheyu Li, Guo Chen, Yanyan Wang, Tao Li
Chinese Medical Journal.2024;[Epub] CrossRef - Liraglutide Attenuates Diabetic Cardiomyopathy via the ILK/PI3K/AKT/PTEN Signaling Pathway in Rats with Streptozotocin-Induced Type 2 Diabetes Mellitus
Shatha M. Alobaid, Rahaf M. Alshahrani, Asma S. Alonazi, Nawal M. Alrasheed, Maha A. Alamin, Tahani K. Alshammari, Anfal F. Bin Dayel, Doaa M. Elnagar, Rana R. Alotaibi, Lama A. Almuthnabi, Dalia H. Almasud, Shahad E. Al-Ammar, Shahad O. Almadhi, Reema A.
Pharmaceuticals.2024; 17(3): 374. CrossRef - An Overview of Diabetic Cardiomyopathy
Abdul Quaiyoom, Ranjeet Kumar
Current Diabetes Reviews.2024;[Epub] CrossRef - Evaluation and Management of Patients With Diabetes and Heart Failure: A Korean Diabetes Association and Korean Society of Heart Failure Consensus Statement
Kyu-Sun Lee, Junghyun Noh, Seong-Mi Park, Kyung Mook Choi, Seok-Min Kang, Kyu-Chang Won, Hyun-Jai Cho, Min Kyong Moon
International Journal of Heart Failure.2023; 5(1): 1. CrossRef - Evaluation and Management of Patients with Diabetes and Heart Failure: A Korean Diabetes Association and Korean Society of Heart Failure Consensus Statement
Kyu-Sun Lee, Junghyun Noh, Seong-Mi Park, Kyung Mook Choi, Seok-Min Kang, Kyu-Chang Won, Hyun-Jai Cho, Min Kyong Moon
Diabetes & Metabolism Journal.2023; 47(1): 10. CrossRef - Machine learning for spatial stratification of progressive cardiovascular dysfunction in a murine model of type 2 diabetes mellitus
Andrya J. Durr, Anna S. Korol, Quincy A. Hathaway, Amina Kunovac, Andrew D. Taylor, Saira Rizwan, Mark V. Pinti, John M. Hollander, Yoshihiro Fukumoto
PLOS ONE.2023; 18(5): e0285512. CrossRef - Hyperglycemic memory in diabetic cardiomyopathy
Jiabing Zhan, Chen Chen, Dao Wen Wang, Huaping Li
Frontiers of Medicine.2022; 16(1): 25. CrossRef - Murine Models of Obesity
Tânia Martins, Catarina Castro-Ribeiro, Sílvia Lemos, Tiago Ferreira, Elisabete Nascimento-Gonçalves, Eduardo Rosa, Paula Alexandra Oliveira, Luís Miguel Antunes
Obesities.2022; 2(2): 127. CrossRef - The Role of Mitochondria in Metabolic Syndrome–Associated Cardiomyopathy
Jiayu Li, Jingye Li, Yijun Chen, Wenyu Hu, Xuhe Gong, Hui Qiu, Hui Chen, Yanguo Xin, Hongwei Li, Tao Li
Oxidative Medicine and Cellular Longevity.2022; 2022: 1. CrossRef - Guidelines on models of diabetic heart disease
Lisa C. Heather, Anne D. Hafstad, Ganesh V. Halade, Romain Harmancey, Kimberley M. Mellor, Paras K. Mishra, Erin E. Mulvihill, Miranda Nabben, Michinari Nakamura, Oliver J. Rider, Matthieu Ruiz, Adam R. Wende, John R. Ussher
American Journal of Physiology-Heart and Circulatory Physiology.2022; 323(1): H176. CrossRef - Extracellular vesicle therapy for non-ischemic heart failure: A systematic review of preclinical studies
Ramana Vaka, Sophie Van Remortel, Valentina Ly, Darryl R. Davis
Extracellular Vesicle.2022; 1: 100009. CrossRef - Effect of a Six-week Endurance Exercise Program and Empagliflozin Consumption on Some Structural and Functional Indices of the Heart in Male Diabetic Rats
Eftekhar Mohammadi, Mohammad Fathi, Farzaneh Chehel Cheraghi, Afshin Nazari
journal of ilam university of medical sciences.2022; 30(3): 1. CrossRef - Cardiac Phosphodiesterases Are Differentially Increased in Diabetic Cardiomyopathy
Rita Hanna, Wared Nour-Eldine, Youakim Saliba, Carole Dagher-Hamalian, Pia Hachem, Pamela Abou-Khalil, Delphine Mika, Audrey Varin, Magali Samia El Hayek, Laëtitia Pereira, Nassim Farès, Grégoire Vandecasteele, Aniella Abi-Gerges
Life Sciences.2021; 283: 119857. CrossRef
 PubReader
PubReader ePub Link
ePub Link Cite
Cite