Inhibition of Serotonin Synthesis Induces Negative Hepatic Lipid Balance
Article information

Abstract
Background
Hepatic steatosis is caused by metabolic stress associated with a positive lipid balance, such as insulin resistance and obesity. Previously we have shown the anti-obesity effects of inhibiting serotonin synthesis, which eventually improved insulin sensitivity and hepatic steatosis. However, it is not clear whether serotonin has direct effect on hepatic lipid accumulation. Here, we showed the possibility of direct action of serotonin on hepatic steatosis.
Methods
Mice were treated with para-chlorophenylalanine (PCPA) or LP-533401 to inhibit serotonin synthesis and fed with high fat diet (HFD) or high carbohydrate diet (HCD) to induce hepatic steatosis. Hepatic triglyceride content and gene expression profiles were analyzed.
Results
Pharmacological and genetic inhibition of serotonin synthesis reduced HFD-induced hepatic lipid accumulation. Furthermore, short-term PCPA treatment prevented HCD-induced hepatic steatosis without affecting glucose tolerance and browning of subcutaneous adipose tissue. Gene expression analysis revealed that the expressions of genes involved in de novo lipogenesis and triacylglycerol synthesis were downregulated by short-term PCPA treatment as well as long-term PCPA treatment.
Conclusion
Short-term inhibition of serotonin synthesis prevented hepatic lipid accumulation without affecting systemic insulin sensitivity and energy expenditure, suggesting the direct steatogenic effect of serotonin in liver.
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) is common liver disease and occurs due to the lipid accumulation in the liver. It is exacerbated from steatosis through steatohepatitis to cirrhosis and hepatocellular carcinoma [12]. NAFLD is closely associated with obesity, which induces positive hepatic lipid balance leading to lipid accumulation and pathological metabolic disturbances in the liver [345]. Because excessive energy intake leads to positive lipid balance and obesity, regulation of systemic energy metabolism is a possible therapeutic approach against NAFLD [678].
Serotonin (5-hydroxytryptamine) is a neurotransmitter involved in the regulation of mood, appetite, and stress responses in the brain [9]. Recently, peripheral serotonin is emerging as a regulator of systemic energy homeostasis. Peripheral serotonin system is functionally separated from central serotonin system because serotonin cannot cross blood brain barrier (BBB) [10]. Serum level of serotonin was elevated in mice fed high fat diet (HFD) [11]. Increasing systemic serotonin activity by knocking out serotonin transporter caused severe obesity, insulin resistance, and hepatic steatosis [1213]. Inhibition of serotonin synthesis in periphery reduced obesity by increasing energy expenditure and decreasing energy storage [1415]. Genetic inhibition of tryptophan hydroxylase 1 (Tph1), a rate-limiting enzyme for serotonin synthesis in periphery, in adipose tissues resulted in the insulin-sensitizing and anti-obesity effects by increasing energy expenditure [14]. Thus, peripheral serotonin works as an obesity hormone which leads to positive energy and lipid balance.
Most of body serotonin is synthesized and secreted from enterochromaffin cells in the gut. Since gut-derived serotonin (GDS) which is released from gut is stored in platelets or metabolized in liver, plasma free serotonin level in peripheral blood is very low. However, considering that liver is the first organ encountering GDS via portal vein and free serotonin levels in portal blood is relatively higher, liver can be the target of GDS. Although inhibition of serotonin synthesis in peripheral tissues reduced hepatic lipid accumulation [15], it has not been tested if serotonin has direct effects on liver. In this study, we explored the functional relationship between hepatic steatosis and serotonin by using pharmacological and genetic models of serotonin inhibition and have shown that serotonin has direct effects on hepatic lipid accumulation.
METHODS
Animal experiments
The experimental protocol for this study was approved by the Institutional Animal Care and Use Committee at the Korea Advanced Institute of Science and Technology (KA2011-29). Tph1 floxed (Tph1fl/fl) mice [16] were crossed with Adiponectin-Cre mice [17] to generate fat-specific Tph1-knockout (Tph1 FKO). 5-Hydroxytryptamine receptor 3A (Htr3a) knockout (KO) mice (B6.129X1-Htr3atm1jul/J) were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). Sequences of primers used in mouse genotyping are given in Table 1. C57BL/6J mice were purchased from the Charles River Japan (Yokohama, Japan). Mice were housed on 12-hour light-dark cycle in climate controlled specific pathogen-free barrier facility. Diet and water were provided ad libitum. At 8 or 12 weeks of age, mice were fed a standard chow diet (SCD), HFD (60% fat calories), or high carbohydrate diet (HCD, 70% carbohydrate calories). Animal diets used in this study were purchased from Research Diets (New Brunswick, NJ, USA). Para-chlorophenylalanine (PCPA, Sigma-Aldrich, St. Louis, MO, USA) was dissolved in phosphate-buffered saline (PBS). PBS or 300 mg/kg PCPA was daily administered by intraperitoneal injection. LP-533401 (Dalton Pharma Services, Toronto, ON, Canada) was dissolved in polyethylene glycol 400 (Sigma-Aldrich) and 5% dextrose (40:60 v/v). Vehicle or 30 mg/kg LP-533401 was administered daily by oral gavage.

Sequences of primers used in mouse genotyping
Histological analysis
Preparation of tissue sections and H&E staining were performed as previously described [1819]. Liver tissues and inguinal white adipose tissues were harvested, fixed in 4% (w/v) paraformaldehyde and embedded in paraffin. Five-µm-thick tissue sections were deparaffinized, rehydrated, and stained with H&E.
Quantification of hepatic triglyceride levels
Liver tissues were homogenized using FastPrep-24 (MP Biomedicals, Santa Ana, CA, USA) in 5% nonyl phenoxypolyethoxylethanol (NP-40; Sigma-Aldrich). Aliquot of homogenates were used for measurement of protein concentrations with BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA). The remaining homogenates were heated to 95℃ for 5 minutes and cooled at room temperature; this cycle was repeated to solubilize all hepatic fat. After centrifugation, supernatants were used for measurement of hepatic triglyceride (TG) levels. Triglyceride Reagent (Sigma-Aldrich) or PBS was added, and samples were incubated at 37℃ for 30 minutes to hydrolyze hepatic TG into glycerol. Samples were then incubated with Free Glycerol Reagent (Sigma-Aldrich) at 37℃ for 5 minutes for colorimetric assay of hydrolyzed TG levels. Differences in absorbance at 540 nm for hydrolyzed or non-hydrolyzed TG were quantified using a glycerol standard. Hepatic TG contents were normalized by protein amounts of liver tissues.
Glucose and insulin tolerance tests
For the glucose tolerance test, overnight-fasted mice were intraperitoneally injected with 2 g/kg D-glucose (Sigma-Aldrich) in PBS. For the insulin tolerance test, 0.75 U/kg human insulin (Humulin R; Lilly, Indianapolis, IN, USA) was intraperitoneally injected into mice after fasting for 6 hours. Blood samples were then obtained from the tail vein at 0, 15, 30, 45, 60, 90, and 120 minutes after injection. Glucose concentrations were measured using a Gluco DR Plus glucometer (Allmedicus, Anyang, Korea).
Quantitative reverse transcription polymerase chain reaction analysis
TRIzol reagent (Ambion, Carlsbard, CA, USA) was used for total RNA extraction from harvested tissues. To eliminate genomic DNA, RNA was treated with TURBO DNase (Ambion). Complementary DNA was generated with Superscript III Reverse Transcriptase (Invitrogen, Carlsbad, CA, USA) from 1 µg of total RNA. Real-time quantitative reverse transcription polymerase chain reaction was performed with Fast SYBR Green Master Mix (Applied Biosystems, Foster City, CA, USA) and a ViiA 7 real-time PCR system (Applied Biosystems) according to the manufacturer's instructions. Expressional profiles were quantified based on the relative delta delta Ct (threshold cycle) method [20] with β-actin as a reference gene. The sequences of primers are given in Table 2.

Sequences of primers used in quantitative real-time polymerase chain reaction
Statistical analysis
Data are presented as the mean±standard error of mean. The values were compared using Student t-test or one-way analysis of variance. Differences with P values of less than 0.05 were considered statistically significant.
RESULTS
Serotonin inhibition protects against HFD-induced hepatic steatosis
In order to determine the effects of serotonin inhibition on diet-induced hepatic steatosis, we used several models of serotonin inhibition on HFD. Pharmacologically, serotonin synthesis can be blocked by Tph inhibitor, PCPA, or LP-533401, which targets the rate-limiting step of serotonin synthesis. Firstly, we examined the livers of mice fed an HFD and treated with PCPA. Histological analysis revealed that HFD-induced hepatic steatosis was blocked by PCPA treatment (Fig. 1A). Accordingly, hepatic TG content was significantly decreased by PCPA treatment (Fig. 1B). PCPA can cross BBB and is known to increase food intake by inhibiting serotonin production in the brain [21]. To further confirm the effects of peripheral serotonin inhibition, we investigated the effect of LP-533401, a peripheral Tph inhibitor which cannot cross the BBB, on hepatic steatosis [22]. Similar to the results with PCPA, LP-533401 also decreased HFD-induced hepatic TG accumulation (Fig. 1C). We also tested Tph1 FKO and Htr3a KO mice, which exhibit increased energy expenditure on HFD [14]. Hepatic TG content was decreased in the liver of both Tph1 FKO and Htr3a KO mice (Fig. 1D and E). Thus, inhibition of peripheral serotonin either by decreased synthesis or reduced activity, protects from HFD-induced hepatic steatosis.

Serotonin inhibition protected against high fat diet (HFD)-induced hepatic steatosis. Eight-week-old mice were fed a standard chow diet (SCD) or HFD for 12 weeks with vehicle, para-chlorophenylalanine (PCPA), or LP-533401 treatment. (A) H&E staining of liver sections from SCD- or HFD-fed mice with vehicle or PCPA treatment. (B) Quantification of hepatic triglyceride (TG) levels in PCPA-treated mice (n=6). (C–E) H&E staining of liver sections (left) and quantification of hepatic TG levels (right) from HFD-fed mice treated with LP-533401 (C), fat-specific Tph1-knockout (Tph1 FKO) mice (D), and 5-hydroxytryptamine receptor 3A (Htr3a) knockout (KO) mice (E) (n=6). Representative images are shown. Scale bars, 50 µm. Tphfl/fl, tryptophan hydroxylase 1 floxed. aP<0.05, bP<0.01, cP<0.001.
PCPA blocks positive hepatic lipid balance on HFD
Hepatic steatosis results from a positive hepatic lipid balance, which occurs through integration of major five components: de novo lipogenesis, TG synthesis, fatty acid (FA) uptake, FA oxidation, and very low density lipoprotein (VLDL) secretion [423]. Each component is again determined by the amount of substrate, allosteric regulation by metabolites, and amount and activity of enzymes of their pathways. To identify the components regulated by serotonin, we investigated gene expression profiles of enzymes involved in hepatic lipid metabolism. PCPA decreased the expression of most genes related to de novo lipogenesis, TG synthesis, and FA uptake (Fig. 2A–C). Thus, PCPA blocks the induction of a positive hepatic lipid balance by HFD. In contrast, the expression of genes involved in FA oxidation and VLDL secretion was not changed by PCPA treatment (Fig. 2D and E). Because most genes in the affected pathway were downregulated, we hypothesized that these changes may reflect the regulation of upstream transcription factors. Indeed, PCPA treatment decreased the expression of lipogenic transcription factors such as Pparg (peroxisome proliferator activated receptor gamma), Nr1h3 (Lxrα; nuclear receptor subfamily 1, group H, member 3 [LXR, liver X receptor]), and Ppargc1a (Pparg coactivator 1 alpha) (Fig. 2F).
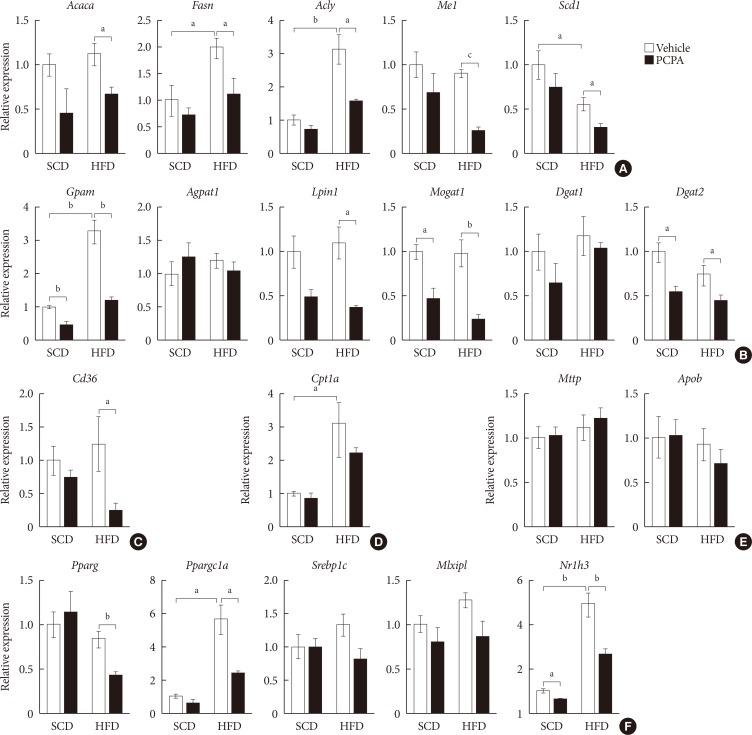
Para-chlorophenylalanine (PCPA) treatment suppressed the positive hepatic lipid balance. Eight-week-old mice were fed a standard chow diet (SCD) or high fat diet (HFD) for 12 weeks and treated with vehicle or PCPA treatment. Hepatic expressional profiles of genes related to de novo lipogenesis (A), triglyceride synthesis (B), fatty acid (FA) uptake (C), FA oxidation (D), very low density lipoprotein secretion (E), and transcription factors (F) were assessed by quantitative reverse transcription polymerase chain reaction (n=6). Acaca, acetyl-CoA carboxylase alpha; Fasn, fatty acid synthase; Acly, ATP citrate lyase; Me1, malic enzyme 1; Scd1, stearoyl-CoA desaturase 1; Gpam, glycerol-3-phosphate acyltransferase; Agpat1, 1-acylglycerol-3-phosphate O-acyltransferase 1; Lpin1, lipin 1; Mogat1, monoacylglycerol O-acyltransferase 1; Dgat1, diacylglycerol O-acyltransferase 1; Dgat2, diacylglycerol O-acyltransferase 2; Cpt1a, carnitine palmitoyltransferase 1a; Mttp, microsomal triglyceride transfer protein; Apob, apolipoprotein B; Pparg, peroxisome proliferator activated receptor gamma; Ppargc1a, Pparg coactivator 1 alpha; Srebp1c, sterol regulatory element binding transcription factor 1c; Mlxipl, MLX interacting protein-like (ChREBP, carbohydrate response element binding protein); Nr1h3, nuclear receptor subfamily 1, group H, member 3 (LXR, liver X receptor). aP<0.05, bP<0.01, cP<0.001.
Short-term intervention of PCPA protects against hepatic steatosis independently from energy expenditure and insulin sensitivity
The alleviation of HFD-induced hepatic steatosis by PCPA could be largely attributed to the decreased flux of steatogenic substrates from adipose tissues resulting from the increased energy expenditure in adipose tissues [1415]. In this case, most of gene expressions in liver of mice fed with HFD were supposed to be similar with those of mice fed with SCD. However, some of the gene expressions were downregulated by PCPA even in liver of mice fed with SCD. These data strongly suggested the direct action of serotonin on liver where several serotonin receptors are expressed [24]. In order to determine the direct effects of serotonin on hepatic steatosis regardless of systemic insulin resistance and energy expenditure, we employed short-term intervention with an HCD and PCPA for 2 weeks. Notably, the short-term HCD had no effect on glucose tolerance (Fig. 3A) or insulin sensitivity (Fig. 3B), and short-term PCPA treatment did not induce beige adipogenesis in inguinal white adipose tissue (Fig. 3C), which is the characteristic of long-term intervention with PCPA [14]. Similar to the HFD model, the HCD induced hepatic TG accumulation, and PCPA decreased hepatic steatosis, resulting in decreased hepatic TG content (Fig. 3D and E).

Short-term treatment with para-chlorophenylalanine (PCPA) in the context of high carbohydrate diet (HCD) protected against hepatic steatosis independently from energy expenditure and insulin sensitivity. Twelve-week-old mice were fed a standard chow diet (SCD) or HCD for 2 weeks and treated with vehicle or PCPA treatment. Intraperitoneal glucose tolerance tests (A) and insulin tolerance tests (B) were performed (n=4). (C) H&E staining of inguinal white adipose tissue sections from HCD-fed mice with vehicle or PCPA treatment. (D) H&E staining of liver sections from SCD- or HCD-fed mice with vehicle or PCPA treatment. (E) Quantification of hepatic triglyceride levels in PCPA-treated mice (n=6). Representative images are shown. Scale bars, 50 µm. aP<0.05.
Short-term intervention of PCPA decreases de novo lipogenesis and TG synthesis
We have shown that the inhibition of hepatic lipid accumulation by PCPA was not entirely attributed to the increased energy expenditure in adipose tissues but to the inhibition of direct action of serotonin on liver. In order to know more precise mechanism of serotonin on hepatic lipid accumulation, we checked gene expression profiles of lipid metabolism in the livers of PCPA-treated mice. Among components of hepatic lipid balance, genes of de novo lipogenesis and TG synthesis were upregulated by HCD, which were reversed by PCPA (Fig. 4A and B), similar to the results in HFD-fed PCPA-treated mice. However, there were no changes in genes encoding components of FA uptake, FA oxidation, and VLDL secretion (Fig. 4C–E). Because lipogenic genes were downregulated, we also investigated upstream transcription factors. Expression levels of Pparg, Srebp1c (sterol regulatory element binding transcription factor 1c), and Mlxipl (MLX interacting protein-like [Chrebp, carbohydrate response element binding protein]), which are lipogenic transcription factors, were significantly decreased (Fig. 4F). Collectively, these data suggested that PCPA induced negative hepatic lipid balance by decreasing de novo lipogenesis and TG synthesis via downregulation of lipogenic transcription factors independently from insulin sensitivity and energy expenditure.
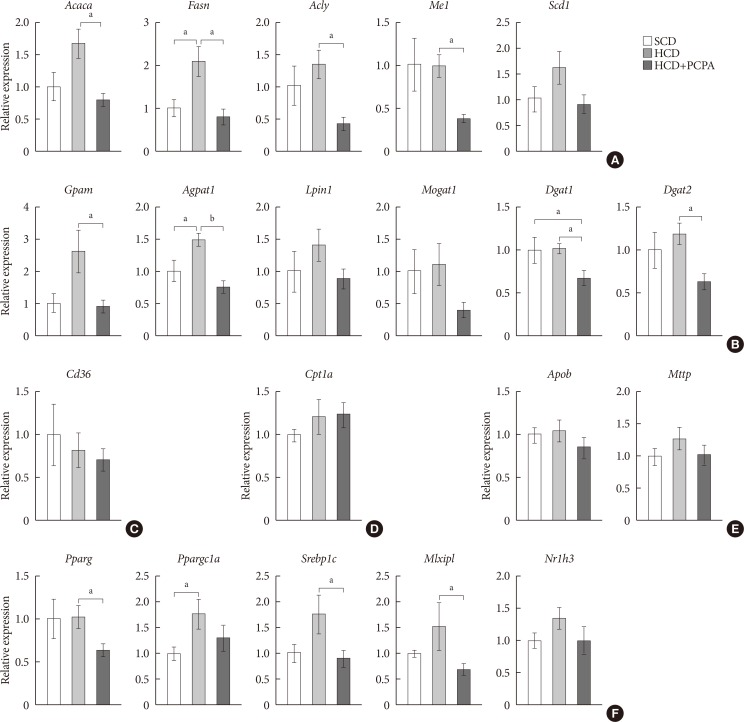
Para-chlorophenylalanine (PCPA) treatment suppressed the positive hepatic lipid balance via downregulation of Pparg, Srebp1c, and Mlxipl. Twelve-week-old mice were fed a standard chow diet (SCD) or high carbohydrate diet (HCD) for 2 weeks with vehicle or PCPA treatment. Hepatic expressional profiles of genes related to de novo lipogenesis (A), triglyceride synthesis (B), fatty acid (FA) uptake (C), FA oxidation (D), very low density lipoprotein secretion (E), and transcription factors (F) were assessed by quantitative reverse transcription polymerase chain reaction (n=6). Acaca, acetyl-CoA carboxylase alpha; Fasn, fatty acid synthase; Acly, ATP citrate lyase; Me1, malic enzyme 1; Scd1, stearoyl-CoA desaturase 1; Gpam, glycerol-3-phosphate acyltransferase; Agpat1, 1-acylglycerol-3-phosphate O-acyltransferase 1; Lpin1, lipin 1; Mogat1, monoacylglycerol O-acyltransferase 1; Dgat1, diacylglycerol O-acyltransferase 1; Dgat2, diacylglycerol O-acyltransferase 2; Cpt1a, carnitine palmitoyltransferase 1a; Apob, apolipoprotein B; Mttp, microsomal triglyceride transfer protein; Pparg, peroxisome proliferator activated receptor gamma; Ppargc1a, Pparg coactivator 1 alpha; Srebp1c, sterol regulatory element binding transcription factor 1c; Mlxipl, MLX interacting protein-like (ChREBP, carbohydrate response element binding protein); Nr1h3, nuclear receptor subfamily 1, group H, member 3 (LXR, liver X receptor). aP<0.05, bP<0.001.
DISCUSSION
HFD, HCD, and ethanol intake are known to induce fatty liver both in human and rodent. These metabolic stresses induce a positive lipid balance in the liver either by increasing de novo lipogenesis and FA uptake or decreasing FA oxidation and VLDL secretion. The alteration of lipid balance can be induced by insulin resistance which induces upregulation of lipogenic gene expressions and increased the flux of steatogenic substrates, free FAs on liver [4]. In this study, we found that pharmacological or genetic inhibition of serotonin production alleviated hepatic steatosis and decreased hepatic TG accumulation in HFD- or HCD-induced hepatic steatosis models.
PCPA is known to irreversibly inhibit Tph, the rate-limiting enzyme in serotonin synthesis, and lead to systemic serotonin depletion [252627]. Serotonin is known to exert its anorexigenic effects via Htr2c in the central nervous system [28]. Thus intracranial injection of PCPA increases food intake and obesity [21]. In contrast, intraperitoneal injection of PCPA exerts anti-obesity effects independently from food intake [14]. Thus, peripheral serotonin system is functionally as well as anatomically separated from central serotonin system on metabolism. We found that both systemic inhibition of serotonin production by PCPA and selective inhibition of peripheral serotonin production by LP-533401 suppressed HFD-induced hepatic fatty changes. Therefore, peripheral serotonin is thought be involved in diet-induced hepatic steatosis.
Gene expression profiles of hepatic lipid metabolism showed that PCPA suppressed the HFD-induced positive hepatic lipid balance by downregulation of genes involved in de novo lipogenesis, TG synthesis, and FA uptake, as in a previous study of white adipose tissue [14]. Because insulin resistance increases hepatic steatosis [29] and PCPA ameliorates insulin resistance in the context of an HFD [14], the anti-steatotic effects of PCPA may reflect both the direct effects of decreased serotonin to hepatic serotonin receptors and the indirect effects on other organs, such as adipose tissue and skeletal muscle. To identify the direct effects of serotonin, we used short-term intervention with PCPA with an HCD. Similar to the results in the HFD-fed model, PCPA could prevent HCD-induced hepatic steatosis. This mechanism was independent from energy expenditure and insulin sensitivity. Thus, serotonin could directly regulate hepatic lipid metabolism.
We found decreased expressions of genes associated with de novo lipogenesis and TG synthesis by short-term intervention with PCPA, including downregulation of Mlxipl (carbohydrate-responsive element-binding protein), a major lipogenic transcription factor that functions in response to high glucose [30], as well as lipogenic Srebp1c and Pparg [31]. Together with other reports that serotonin increased fat content in primary hepatocytes [3233], our data strongly suggested that serotonin can be a direct lipogenic stimulus in the liver via regulation of lipogenic transcription factors, as has been observed in adipose tissue. Although causative serotonin receptor and its signaling pathway are not identified, we show the direct action of serotonin on hepatic lipid metabolism. Models of serotonin inhibition showed anti-steatogenic effects on diet-induced hepatic steatosis (Fig. 1), but these are sum of direct effects from decreased hepatic serotonin action and indirect effects from increased energy expenditure and enhanced insulin sensitivity. We tried to distinguish those two mechanisms, and found that short-term intervention of PCPA treatment on HCD didn't change energy expenditure and insulin sensitivity. Thus we firstly identified direct effects of serotonin inhibition on liver in vivo. Additional studies using liver-specific serotonin receptor knockout are required to elucidate the detailed mechanism how serotonin signaling is associated with diet-induced hepatic steatosis. Selective modulation of serotonin pathway might introduce new therapeutic approach against hepatic steatosis.
ACKNOWLEDGMENTS
We thank all the members of the Integrated Lab of Metabolism, Obesity, and Diabetes (iMOD) at KAIST for their helpful discussions and technical support. This work was supported by a grant from the Korean Diabetes Association (grant number: 2017S-1 to Jun Namkung) and grants from the National Research Foundation of Korea (NRF) funded by Ministry of Science, ICT & Future Planning (grant numbers: NRF-2014M3A9D8034464 to Hail Kim and NRF-2016R1C1B1011688 to Jun Namkung).
Notes
CONFLICTS OF INTEREST: No potential conflict of interest relevant to this article was reported.

