Diabetes Promotes Myocardial Fibrosis via AMPK/EZH2/PPAR-γ Signaling Pathway
Article information

Abstract
Background
Diabetes-induced cardiac fibrosis is one of the main mechanisms of diabetic cardiomyopathy. As a common histone methyltransferase, enhancer of zeste homolog 2 (EZH2) has been implicated in fibrosis progression in multiple organs. However, the mechanism of EZH2 in diabetic myocardial fibrosis has not been clarified.
Methods
In the current study, rat and mouse diabetic model were established, the left ventricular function of rat and mouse were evaluated by echocardiography and the fibrosis of rat ventricle was evaluated by Masson staining. Primary rat ventricular fibroblasts were cultured and stimulated with high glucose (HG) in vitro. The expression of histone H3 lysine 27 (H3K27) trimethylation, EZH2, and myocardial fibrosis proteins were assayed.
Results
In STZ-induced diabetic ventricular tissues and HG-induced primary ventricular fibroblasts in vitro, H3K27 trimethylation was increased and the phosphorylation of EZH2 was reduced. Inhibition of EZH2 with GSK126 suppressed the activation, differentiation, and migration of cardiac fibroblasts as well as the overexpression of the fibrotic proteins induced by HG. Mechanical study demonstrated that HG reduced phosphorylation of EZH2 on Thr311 by inactivating AMP-activated protein kinase (AMPK), which transcriptionally inhibited peroxisome proliferator-activated receptor γ (PPAR-γ) expression to promote the fibroblasts activation and differentiation.
Conclusion
Our data revealed an AMPK/EZH2/PPAR-γ signal pathway is involved in HG-induced cardiac fibrosis.
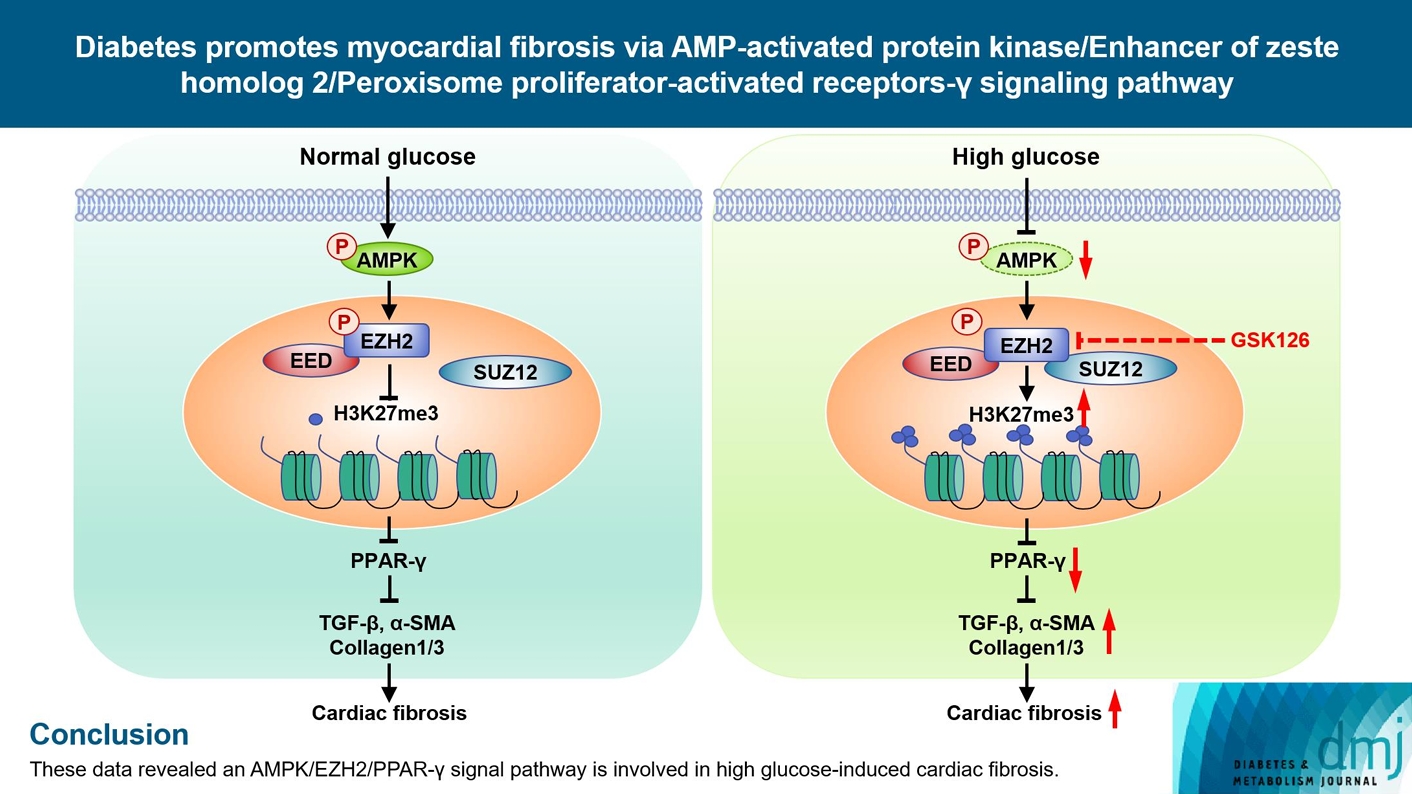
Highlights
• EZH2 was activated in diabetic heart and high glucose-stimulated myofibroblasts.
• High glucose inhibited AMPK-mediated phosphorylation of EZH2.
• Reduced EZH2 phosphorylation inhibited PPAR-γ transcription.
• Inhibition of EZH2 reduced the conversion of fibroblasts into myofibroblasts.
INTRODUCTION
Diabetic cardiomyopathy is a common type of diabetic cardiovascular complications leading to heart failure [1]. During the development of diabetic heart failure, myocardial fibrosis plays an important role in patients with diabetes mellitus (DM) and animal models of diabetes [2,3]. Following injury or stimulus, cardiac fibroblasts (CFs) are activated and transformed into myofibroblasts, which are not present in normal hearts [4]. Myofibroblasts display acerbated proliferative, migratory and contractile abilities with excessive deposition of extracellular matrix (ECM) [5,6]. Overproduction of ECM is involved in cardiac remodeling, which reduces tissue compliance and promotes myocardial fibrosis [7]. Expression of α-smooth muscle actin (α-SMA) is a hallmark of fibroblast activation. However, the mechanism of CFs activation in diabetic condition has not been clarified [8].
Histone methylation contributes to the development of physiologic cardiac fibrosis and heart failure [9]. Polycomb repressive complex 2 (PRC2) as an only identified methyltransferase catalyzing mono-, di-, and trimethylation of lysine 27 on histone H3 (H3K27me3), formed by core subunits embryonic ectoderm development, suppressor of zest 12 (SUZ12), and enhancer of zeste homolog 2 (EZH2) [10]. EZH2, a primary enzymatic catalytic subunit of PRC2, acts as a histone methyltransferase and suppresses target gene transcription by catalyzing H3K27me3 in a PRC2 dependent way [10-12]. EZH2 plays an important role in fibrosis of liver, lung and skin in previous study [13-15]. However, the role of EZH2 in diabetic cardiac fibrosis and myofibroblasts differentiation are still unclear.
It has been reported that EZH2 could be phosphorylated by AMP-activated protein kinase (AMPK) at Thr311 during sustained energy starvation. Phosphorylation of Thr311 prevents EZH2 from interacting with SUZ12, and leads to decreased H3K27me3 [12]. AMPK is a known regulator of diabetic cardiac metabolism [16]. It is activated by phosphorylation of threonine 172 (Thr172) at α subunit and inactivated by declined phosphorylation of Thr172 in diabetic myocardium [17,18]. Inhibition of Peroxisome proliferator-activated receptor γ (PPAR-γ) has been shown to promote the activation and differentiation of myocardial fibroblasts [19]. Previous studies showed that the activation of PPAR-γ inhibited CFs proliferation, ECM excessive deposition and myofibroblasts transformation in angiotensin II (Ang II) treated CFs [20-23].
In the current study, we hypothesized that high glucose (HG) promoted fibroblast differentiation and induced diabetic myocardial fibrosis through EZH2. To clarify this hypothesis, we established diabetic animal model and hyperglycemic stimulated cell model. Our data demonstrated that AMPK/EZH2/PPAR-γ signal pathway played an important role in the development of diabetic cardiac fibrosis.
METHODS
Induction of diabetic model
Sprague-Dawley rats (6 to 8 weeks old, 150 to 200 g, male) and C57BL/6J mice (6 to 8 weeks old, 20 to 25 g, male) were purchased from Changzhou Cavens Laboratory Animal Co. Ltd. in Changzhou, China. The rats and mice were housed under specific pathogen-free conditions with standard food and water. All animals were maintained up to five littermates per cage under a 12-hour light/dark cycle. The rat diabetic model was established as previously described [24]. In brief, the rats were intraperitoneally injected with one dose of streptozotocin (STZ; Sigma-Aldrich Corp., St. Louis, MO, USA; 60 mg/kg). The mouse diabetic model was established via intraperitoneally injected with STZ (55 mg/kg) for 5 continuous days. One week later, rats or mice with a blood glucose concentration >16.7 mmol/L after fasting for 6 hours, were enrolled in this study. The heart specimens were harvested 12 weeks after STZ infusion.
Primary CF culture and treatment
Primary CFs were isolated from the ventricular of neonatal rats (purchased from Changzhou Cavens Laboratory Animal Co. Ltd.). Hearts from neonatal rats were completely minced and digested with 0.125% trypsin (Gibco, Grand Island, NY, USA; 25200072) and 0.1% collagenase (Worthington-Biochem, Lakewood, NJ, USA; LS004176) at 37°C. The digested tissue was then prepared into single cell suspension, and plated in low glucose Dulbecco’s Modified Essential Medium (DMEM, Gibco; 11885092) with 10% fetal bovine serum (FBS; AusgeneX, Molendinar, Australia; FBS500-s) and 1% penicillin/streptomycin (Gibco; 15070063). One hour later, the unattached cells were removed and new complete medium was added. Cells were transferred to 12-well plates with minimal changes in phenotype associated with culture for subsequent experiments. Before treatment, cells were starved in serum-free medium for 24 hours and then treated with HG (Sigma-Aldrich; G7528-250G, 30 mM), competitive inhibitor of PRC2 (GSK126; MedChemExpress, Monmouth Junction, NJ, USA; HY-13470, 500 nM), A769662 (MedChemExpress; HY-50662, 10 μM), or rosiglitazone (Sigma-Aldrich; R2408, 5 μM) for 72 hours.
Left ventricle ejection fraction measurement
Echocardiography was used to evaluate heart function of rats (Philips, New Bedford, MA, USA; IE33) and mice (GE, Chicago, IL, USA; E95). Anesthesia induction of rats was performed with isoflurane and maintained with chloroform. M-mode tracings were used to measure left ventricular fractional shortening (LVFS) and the left ventricular ejection fraction (LVEF). All measurements were made by an observer who was blinded with respect to the identity of the tracings. All measurements were performed over six consecutive cardiac cycles.
Histology
The heart tissues were fixed with 4% paraformaldehyde (Beyotime, Shanghai, China; P0099) for 24 hours after harvest. Then, heart tissues were embedded in paraffin and cut into 5 μm sections. Masson staining was performed and the sections were photographed under microscope.
Cellular immunofluorescence
Primary CFs were seeded into 12-well plates with lysine-coated slides and exposed to HG, GSK126, A769662, or rosiglitazone for 72 hours. The cells were fixed with 4% paraformaldehyde, permeabilized with 0.1% (vol/vol) Triton X-100 (Beyotime; P0096) and blocked with 2% bovine serum albumin (Absin Bioscience Inc., Shanghai, China; abs49001012a).Then the cells were incubated with anti-α-SMA (Abcam, Cambridge, UK; a5694) antibody at 4°C overnight, washed with phosphate buffered solution containing Tween 80 for three times and incubated with the fluorescein-AffiniPure goat anti-rabbit antibody (Jackson ImmunoResearch, West Grove, PA, USA; 111-095-003) and 4´,6-diamidino-2-phenylindole (DAPI, Sigma-Aldrich; F6057-20 mL) at room temperature for 1 hour. Finally, the cells were observed and photographed under a confocal microscope (Leica Microsystems, Wetzlar, Germany).
Transwell migration assay
Primary CFs were stimulated with HG, GSK126, A769662, or rosiglitazone for 72 hours, digested with 0.25% trypsin (Gibco; 25200072) and resuspended with serum-free DMEM. Then these cells were seeded into the cell culture inserts and cultured in DMEM with 20% FBS to the wells of the BD Falcon TC Companion Plate (Costar, Richmond, VA, USA; R3422). After 24 hours, the inside cells of the inset were washed out and remaining cells were fixed in 4% paraformaldehyde for 15 minutes and stained with 0.5% crystal violet for 15 minutes. After drying at room temperature, the cells were observed and counted under microscope.
Scratching assay
Primary CFs were cultured in the six-well plates to a continuity of 90%. The cells were stimulated with HG, GSK126, A769662, or rosiglitazone for 72 hours. Then a sterile 200 μL pipette tip was used to scratch the bottom of the six-well plate. The cells were preceded to culture in serum-free medium for 72 hours, observed and photographed under a microscope.
Quantitative real-time polymerase chain reaction
Total RNA was extracted from neonatal rat CFs or rat ventricular tissues using Trizol reagent (Ambion, Austin, TX, USA; 155 96018). Total RNA was reversely transcribed into cDNA using the PrimeScript RT Master Mix (TAKARA, Kusatsu, Japan; RR036A-1). Quantitative polymerase chain reaction (PCR) was performed using a SYBR Green PCR mix (Roche Co., Basel, Switzerland; 4913914001) on an ABI Prism 7500HT sequence detection system (Applied Biosystems Fisher Scientific Inc., Waltham, MA, USA). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as an internal control to normalize the relative expression of target genes. The threshold cycle (Ct) was determined and the method 2-ΔΔCt was used to calculate the quantitative expression of target mRNA. The sequences of the primers (synthesized by Sangon Biotech, Shanghai, China) for the target genes as followed: EZH2 (rat): forward primer, TTCCAGCACAAGTCATCCCG, reverse primer, GTGCCATCCTGATCCAGAACT; PPAR-γ (rat): forward primer, CGGTTTCAGAAGTGCCTTGC, reverse primer, AAATGCTTTGCCAGGGCTC; GAPDH (rat): forward primer, CGGACGGAAATGCTATGGA, reverse primer, AGCCTGGAAGATGTCGTTGG.
Western blotting analysis
The ventricular tissue and cultured cell were lysed in an ice-cold radioimmunoprecipitation assay (RIPA) buffer (Pierce Co., Rockford, IL, USA; 89901) containing protease and phosphatase inhibitors (Roche Co.; 04693159001). The lysates were fractionated using sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride (PVDF) membranes (MILLEX GV, Merck Millipore, Burlington, MA, USA; SLGV033RB-1). The PVDF membrane was incubated with specific primary antibodies: GAPDH (Cell Signaling Technology Co., Danvers, MA, USA; 2118), EZH2 (Cell Signaling Technology Co.; 5246), phosph-EZH2 (Thr311; Cell Signaling Technology Co.; 27888), H3K27me3 (Cell Signaling Technology Co.; 9733), phosph-AMPK (T172; Cell Signaling Technology Co.; 2535), AMPK (Cell Signaling Technology Co.; 2532), transforming growth factor-β1 (TGF-β1; Abcam; ab1796 95), collagen 1 (Protech, Rosemont, IL, USA; 14695-1-AP), and α-SMA (Abcam; a5694). ImageJ software (Scion Corp., Chicago, IL, USA) was used to quantified the densitometry of immunoblot bands. The GAPDH was used as an internal control.
Statistical analysis
Quantitative data are presented as mean±standard error of the mean. Statistical analysis was performed by SPSS version 22.0 software (IBM Co., Armonk, NY, USA). Levene’s test was used to check the homogeneity of variance. Normally distributed data were compared using the independent Student’s t-test and non-normally distributed data were compared using the Mann-Whitney U-test. A one-way analysis of variance was used to assess significance in four groups for normally distributed data and least significant difference post hoc multiple comparisons were used to assess the significance between two groups. P<0.05 was considered to be statistically significant.
Ethical statement
All animal experiments were conducted following the national guidelines and the relevant national laws on the protection of animals.
RESULTS
H3K27 trimethylation was up-regulated in left ventricle of STZ-induced diabetic rats and mice
To explore the role of histone H3 lysine 27 (H3K27) trimethylation in diabetic myocardial fibrosis, we established a STZ-induced diabetic model in rats. The echocardiographic data were shown that the LVFS and LVEF were decreased in the DM group than that in control group (Fig. 1A), suggesting that the systolic functions of the left ventricle were decreased in STZ-induced diabetic rats. Masson’s trichrome staining demonstrated that the collagen deposition and cardiac fibrosis were increased in the diabetic ventricular tissues (Fig. 1B). Meanwhile, the protein expression of TGF-β1 and α-SMA were higher in DM group than that in control group (Fig. 1C), suggesting that the transdifferentiation of CFs into myofibroblasts occurred in STZ-induced diabetic rats. The level of H3K27me3 and the phosphorylation of EZH2 at T311 were assessed in STZ-induced diabetic rat ventricular tissue. The data were shown that the level of H3K27me3 was higher in diabetic rat than that in control rat. The expression of EZH2 on mRNA and protein level was not significantly increased in the diabetic group compared to control group. However, the phosphorylation of EZH2 at T311 was lower in diabetic rat than that in control rat (Fig. 1D and E). To confirm the alteration of H3K27me3 in diabetic condition, STZ-induced diabetic mice model was established, LVFS and LVEF were decreased in diabetic mice compared to control mice (Fig. 1F). The level of H3K27me3 was higher in diabetic mice than that in control mice. And the protein level of EZH2 was not significantly increased (Fig. 1G).

Histone H3 lysine 27 trimethylation (H3K27me3) was up-regulated in left ventricle of streptozotocin (STZ)-induced diabetic rats and mice. (A) Left ventricular ejection fraction (LVEF) and fractional shortening (FS) were measured by echocardiography in the diabetic and non-diabetic group rats (n=4 in each group). (B) Mason staining of ventricular tissues obtained from nondiabetic and diabetic rats (scale bar 300 μm). (C) Protein levels of extracellular matrix-related genes and α-smooth muscle actin (α-SMA) were evaluated by Western blot in rat ventricular tissues (n=3 in each group). (D) Western blot analysis of the expression of phospho-enhancer of zeste homolog 2 (p-EZH2), EZH2, and H3K27me3 in ventricular tissues from non-diabetic and diabetic rats (n=3 in each group). (E) EZH2 mRNA levels were measured in non-diabetic and diabetic rat ventricular tissues (n=4 in each group). (F) LVEF and FS were measured by echocardiography in the diabetic and non-diabetic group mice (n=6 in each group). (G) Western blot analysis of the expression of EZH2 and H3K27me3 in ventricular tissues from non-diabetic and diabetic mice (n=6 in each group). Data are presented as mean±standard error of the mean. TGF-β, transforming growth factor-β; GAPDH, glyceraldehyde 3-phosphate dehydrogenase. aP<0.05, bP<0.01.
HG promoted the transformation of CFs into myofibroblasts
To determine whether HG induce cardiac fibrosis, CFs were treated with either 5.5mmol/L normal glucose (NG) or 30mmol/L (HG) for 72 hours. Western blotting analysis revealed that ECM proteins such as collagen 1, α-SMA, and TGF-β1, were significantly elevated in HG-treated CFs (Fig. 2A). Immunofluorescence analysis demonstrated that α-SMA was up-regulated with HG treatment in CFs, suggesting that HG promoted activation and differentiation of CFs (Fig. 2B). To explore whether HG promotes migration of CFs, transwell assay and scratchwound healing assay were performed. Transwell data was shown that more migrated CFs treated by HG compared to NG treatment (Fig. 2C). In addition, scratch-wound healing assay data demonstrated that the number and area of cell migration stimulated by HG were higher than those in NG group, suggesting HG promote migration of fibroblast (Fig. 2D). These data suggested that HG promote the transformation of CFs into myofibroblasts.

High glucose (HG) promoted the transformation of cardiac fibroblasts (CFs) into myofibroblasts and reduced the phosphorylation of enhancer of zeste homolog 2 (EZH2) at T311. (A) Protein levels of extracellular matrix-related genes and α-smooth muscle actin (α-SMA) were analyzed in primary CFs after HG treatment (30 mM) for 72 hours (n=3 in each group). (B) Representative pictures of immunofluorescence of α-SMA expression in CFs (red, α-SMA; blue, 4´,6-diamidino-2-phenylindole [DAPI]; scale bar 50 μm). (C) Transwell assays were performed on CFs under HG treatment for 72 hours (scale bar 200 μm, n=3 in each group). (D) Scratching tests were performed on CFs under HG treatment for 72 hours (scale bar 400 μm, n=4 in each group). (E) Western blot analysis of the expression of phospho-EZH2 (p-EZH2), EZH2, and histone H3 lysine 27 trimethylation (H3K27me3) in CFs under HG treatment for 72 hours (n=3 in each group). Data are presented as mean±standard error of the mean. TGF-β, transforming growth factor-β; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; NG, normal glucose. aP<0.05, bP<0.01.
HG reduced the phosphorylation of EZH2 at T311 in CFs
To investigate whether HG induce up-regulation of H3K27me3, CFs were treated with either NG or HG for 72 hours. Consistent with the data in vivo, the level of H3K27me3 were elevated in HG-treated fibroblasts (Fig. 2E), while the protein level of EZH2 were not significantly increased in HG-treated fibroblasts (Fig. 2E). As phosphorylation of EZH2 was involved in regulation of H3K27me3, we investigated the level of phosphorylated EZH2. As shown in Fig. 2E, the phosphorylation of EZH2 at T311 reduced in HG-treated CFs. These data suggested that HG up-regulates H3K27me3 and decrease EZH2 phosphorylation in CFs.
Inhibition of EZH2 attenuated HG-induced CF differentiation
To determine whether HG promotes CF differentiation via EZH2, GSK126, an inhibitor of EZH2 methyltransferase activity, was used to block EZH2 activity in myocardial fibroblasts. After 72 hours incubation with GSK126, the level H3K27me3 was significantly reduced as compared to the cells without GSK126 treatment. And the phosphorylation of EZH2 at T311 was significantly increased as compared to the cells without GSK126 treatment (Fig. 3A). Immunofluorescence analysis showed that GSK126 inhibited the expression of α-SMA in CFs (Fig. 3B), suggesting that HG promoted CF differentiation via EZH2. Furthermore, collagen 1, TGF-β1, and α-SMA expression were down-regulated in CFs induced by HG in the presence of GSK126 (Fig. 3C). Transwell and scratch-wound healing assays revealed that GSK126-treated cells displayed a significant reduction of migration stimulated by HG (Fig. 3D and E). In addition, cell counting kit 8 (CCK-8) assay demonstrated that GSK126 inhibited the proliferation of fibroblasts induced by HG (Fig. 3F). These data suggested that the differentiation of CFs induced by HG might be due to the activation of EZH2.

Enhancer of zeste homolog 2 (EZH2) inhibition attenuated high glucose (HG)-induced fibroblast differentiation and extracellular matrix (ECM) protein synthesis. (A) Western blot analysis of the expression of phospho-EZH2 (p-EZH2), EZH2, and histone H3 lysine 27 trimethylation (H3K27me3) in cardiac fibroblasts (CFs) after HG and GSK126 (500 nM) treatment (n=3 in each group). (B) Inhibition of EZH2 with GSK126 in CFs and Western blot analysis of protein levels of ECM-related genes and α-smooth muscle actin (α-SMA; n=3 in each group). (C) Representative images of immunofluorescence images of α-SMA expression in CFs with different treatment (red, α-SMA; blue, 4´,6-diamidino-2-phenylindole [DAPI]; scale bar 50 μm). (D) Inhibition of EZH2 with GSK126 under HG treatment for 72 hours, transwell assay was performed (scale bar 200 μm, n=3 in each group). (E) Inhibiting of EZH2 with GSK126 under HG treatment for 72 hours, scratching tests was performed (scale bar 400 μm, n=8 in each group). (F) CFs proliferation were measured with cell counting kit 8 (CCK-8) under HG treatment of for 72 hours with/without GSK126 (n=3 in each group). Data are presented as mean±standard error of the mean. GAPDH, glyceraldehyde 3-phosphate dehydrogenase; TGF-β, transforming growth factor-β; NG, normal glucose; DMSO, dimethyl sulfoxide; OD, optical density. aP< 0.05, bP<0.01.
EZH2 regulated cardiac fibrosis through the PPAR-γ/TGF-β1 signaling pathway
To explore the potential mechanism by which EZH2 promotes cardiac fibrosis under HG condition, PPAR-γ was examined in myocardial fibroblasts stimulated with HG. The data was shown that PPAR-γ transcription was reduced in HG-induced myocardial fibroblasts, while the reduction of PPAR-γ was recovered by GSK126 treatment (Fig. 4A). Rosiglitazone, an agonist for PPAR-γ, inhibited the expression of α-SMA, TGF-β1, and collagen 1 in CFs induced by HG (Fig. 4B). Immunofluorescence analysis also demonstrated that rosiglitazone inhibits expression of α-SMA in CF treated with HG (Fig. 4C). Furthermore, migration of CFs induced by HG was suppressed in the presence of rosiglitazone through transwell and scratch-wound healing assays (Fig. 4D and E). Overall, these data suggested that HG might induce cardiac fibrosis via EZH2/PPAR-γ/TGF-β1 signal pathway.
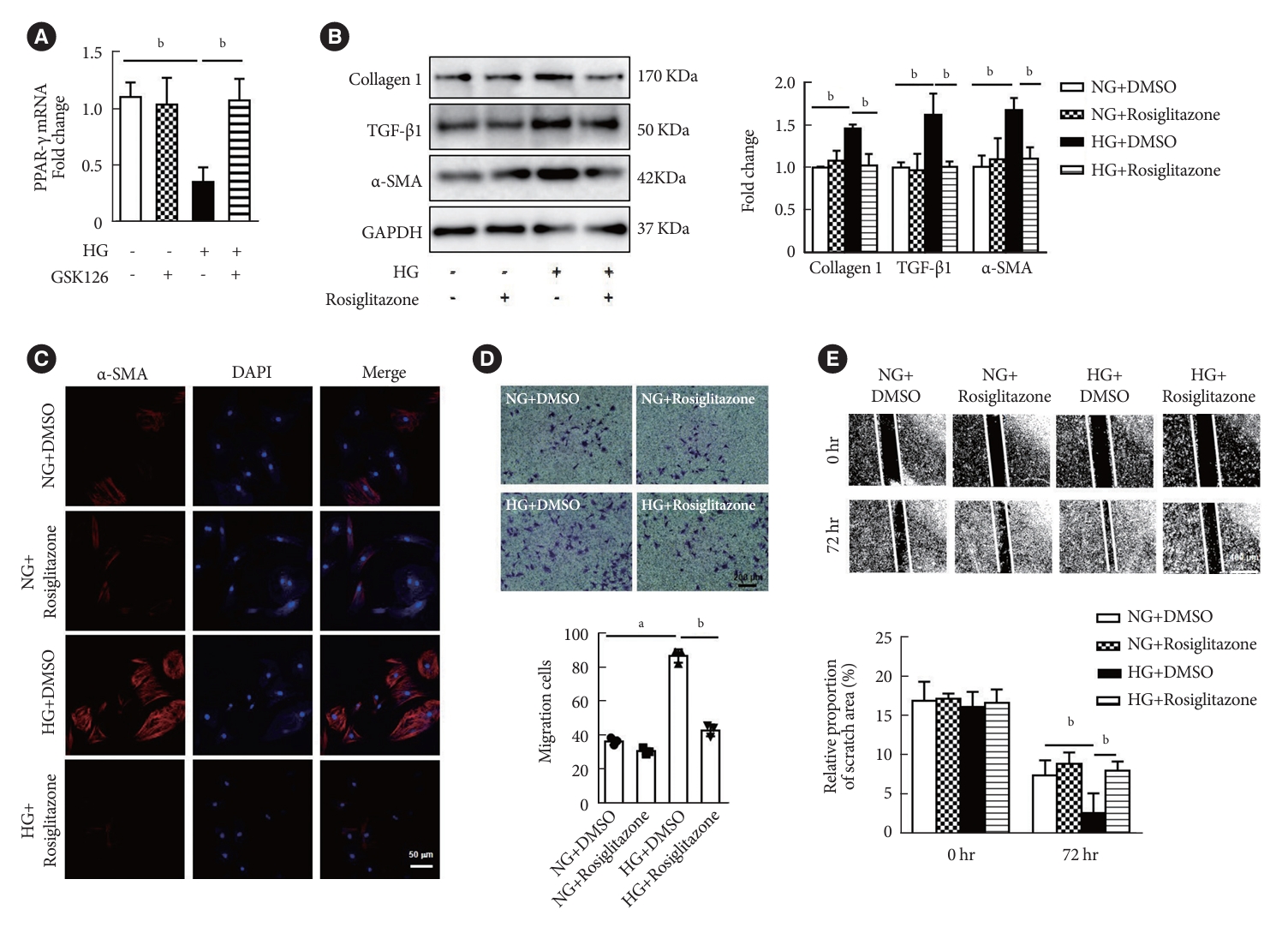
Peroxisome proliferator-activated receptor γ (PPAR-γ) activation attenuated high glucose (HG)-induced fibroblasts differentiation and extracellular matrix (ECM) proteins synthesis. (A) PPAR-γ mRNA levels were measured in cardiac fibroblasts (CFs) after HG and GSK126 (500 nM, n=3 in each group). (B) Activation of PPAR-γ with rosiglitazone (50 μM) in CFs and Western blot analysis of protein levels of ECM-related genes and α-smooth muscle actin (α-SMA; n=3 in each group). (C) Immunofluorescence images of α-SMA expression in CFs with different treatment (red, α-SMA; blue, 4´,6-diamidino-2-phenylindole [DAPI]; scale bar 50 μm). (D) Activation of PPAR-γ with rosiglitone under HG treatment for 72 hours, transwell assay was performed (scale bar 200 μm, n=3 in each group). (E) Activation of PPAR-γ with rosiglitone under HG treatment for 72 hours (scale bar 400 μm, n=6 in each group). Data are presented as mean±standard error of the mean. TGF-β, transforming growth factor-β; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; NG, normal glucose; DMSO, dimethyl sulfoxide. aP<0.05, bP<0.01.
AMPK suppressed EZH2-mediated H3K27me3 by phosphorylating EZH2
To explore the mechanism that HG activates EZH2, the expression of AMPK was examined in HG condition. Our data was shown that phosphorylation of AMPK was decreased in diabetic rats and HG-treated CFs (Fig. 5A and B). A specific AMPK agonist, A769662, was added to CFs treated with HG. The data were shown that A769662 treatment led to phosphorylation of EZH2 at T311 restored, and inhibited the expression of H3K27me3 induced by HG (Fig. 5C). The expressions of phospho-AMPK (p-AMPK), AMPK, p-EZH2, EZH2, and H3K27me3 were examined in myocardial tissues of diabetic mice treated with AMPK agonist A769662. In myocardial tissue of diabetic mice treated with A769662, the phosphorylation EZH2 and AMPK were increased as compared to the myocardial tissue of diabetic mice without A769662 treatment. And H3K27me3 was reduced as compared to the myocardial tissue of diabetic mice without A769662 treatment (Fig. 5D). These results suggested that activation of AMPK suppressed EZH2-mediated H3K27me3 by phosphorylating EZH2.

Activation of AMP-activated protein kinase (AMPK) promoted enhancer of zeste homolog 2 (EZH2) T311 phosphorylation. (A) Western blot analysis the expression of phospho-AMPK (p-AMPK) and AMPK in cardiac fibroblasts (CFs) under high glucose (HG) treatment for 72 hours (n=3 in each group). (B) Western blot analysis of the expression of p-AMPK and AMPK in tat myocardial tissue with diabetic (n=3 in each group). (C) Western blot analysis of the expression of p-AMPK, AMPK, p-EZH2, and EZH2 in CFs after HG and activator of AMPK (A769662, 10 μM) treatment (n=3 in each group). (D) Western blot analysis of the expression of p-AMPK, AMPK, p-EZH2, EZH2, and histone H3 lysine 27 trimethylation (H3K27me3) in myocardial tissues of diabetic mice treated with AMPK agonist A769662 (n=3 in each group). Data are presented as mean±standard error of the mean. NG, normal glucose; t-AMPK, total AMPK; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; DMSO, dimethyl sulfoxide. aP<0.05, bP<0.01.
DISCUSSION
A small but compelling body of evidence has accumulated in recent years implicating EZH2 in organ fibrosis. However, the role of EZH2 in diabetic cardiac fibrosis and myofibroblasts differentiation are still unclear. In the present study, our data showed that: (1) activation of EZH2 and H3K27me3 were increased in STZ-induced diabetic rat cardiac and mice cardiac, as well as in cultured fibroblasts stimulated by HG; (2) inhibition of EZH2 reduced the proliferation, migration, conversion of CFs and excessive ECM protein deposition produced by CFs in response to HG; (3) AMPK-mediated EZH2 phosphorylation regulates the PPAR-γ/TGF-β1 signaling pathway via histone methylation under HG stimulation (Fig. 6).
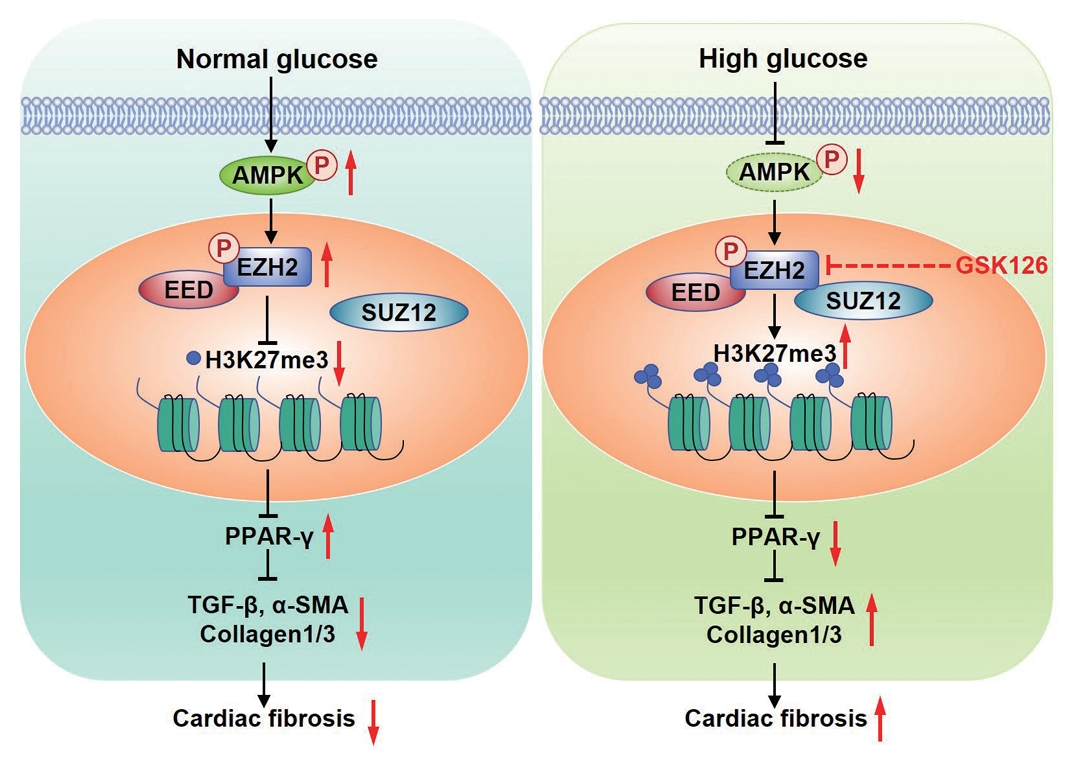
The mechanism of diabetes promoting myocardial fibrosis via AMP-activated protein kinase (AMPK)/enhancer of zeste homolog 2 (EZH2)/peroxisome proliferator-activated receptor γ (PPAR-γ) signaling pathway. Under normal glucose, phosphorylation of AMPK phosphorylates EZH2, which inhibits the trimethylation activity EZH2, leading to expression of PPAR-γ and inhibition of myocardial fibroblasts activation. Under the condition of diabetics, high glucose inactivates AMPK, increases trimethylation activity EZH2 by reducing the phosphorylation of EZH2 at T311, which represses transcription of PPAR-γ and suppressed diabetic fibrosis. P, phosphorylation; EED, embryonic ectoderm development; SUZ12, suppressor of zest 12; H3K27me3, histone H3 lysine 27 trimethylation; TGF-β, transforming growth factor-β; α-SMA, α-smooth muscle actin; GSK126, competitive inhibitor of PRC2.
The expression of EZH2 in cardiac tissue induced by STZ and CFs treated with HG
EZH2 is the catalytic subunit of PRC2 which primary catalytic methylation of H3K27. Previous studies have report that EZH2 is involved in cardiac hypertrophy, cardiac differentiation of human embryonic stem, myocardial apoptosis, proliferation, regeneration, atrial fibrillation, and ion channel disorder [25-31]. EZH2 is also involved in the progression of atherosclerosis [32]. The role of EZH2 was focused to apoptosis, proliferation, and hypertrophy of cardiomyocytes or differentiation of embryonic stem cells. However, the role of EZH2 in CFs is still unclarified. Our study demonstrated that H3K27me3 was significantly up-regulated in cardiac tissue of diabetic rats and mice, which was consistent with the previews study [33]. However, the expression of EZH2 was observed no significant change in both HG-treated CFs and STZ-induced cardiac tissues compared with control group. While phosphorylation of EZH2 at T311 reduced significantly in cardiac tissue of diabetic rats and HG-treated CFs. In the previews study, the expression of EZH2 and H3K27me3 increased spontaneously in diabetic (db/db) C57BL/Ks mice and HG-cultured mouse cardiomyocytes. The animal model included in this study was type 2 diabetes mellitus mouse, while the cells were myocardial cells. And our animal model was STZ-induced type 1 diabetes mellitus, while the cell was CFs, which suggested that the role of EZH2 in myocardial pathological changes induced by type 1 diabetes mellitus and type 2 diabetes mellitus might be different between myocardial cells and CFs.
Role of EZH2 in diabetic cardiac fibrosis and CFs differentiation
Studies have shown that EZH2 plays an important role in organ fibrosis. Tsou et al. [13] provided support for the involvement of EZH2 in the expression of profibrotic genes including collagen type I-alpha 1 (COL1A1), fos-related antigen-2 (FRA2), and TGF-β1. EZH2 inhibitor decreased migratory and contractility abilities in scleroderma dermal fibroblasts. EZH2 was activated in fibroblast in the lungs of patients with idiopathic pulmonary fibrosis and bleomycin-challenged mice in vivo via increasing the p-Smad2/3-TGF-β1 signaling pathway in fibroblasts in vitro [34].
EZH2 has been shown to induce atrial fibrosis by regulating atrial CFs in atrial fibrillation [30], and contribute to the expression of fibrosis-associated genes stimulated by Ang II in atrial myofibroblasts [35]. However, the role of EZH2 in diabetic ventricular fibrosis is still unclear. In our study, we found that EZH2 promoted ventricular fibroblast differentiation and led to ventricular fibrosis. Myocardial fibrosis is characteristics as excessive ECM protein deposition [36], which mainly synthesized by the activated myofibroblasts. After treatment of EZH2 inhibitor, GSK126, HG-induced expression of α-SMA, TGF-β1, and collagen 1 was decreased, suggesting that EZH2 activity was critical for activation and differentiation of CFs. Increased migration and proliferation were another marker of CF activation and promotion of fibrosis [37]. GSK126 treatment reduced migration and proliferation of CFs with HG stimulation. TGF-β1 is a well-known key cytokine that promotes fibrosis and CF differentiation [6,38]. The expression of TGF-β1 was reduced in response to GSK126 intervention in HG-treated CFs. Our results suggested that EZH2 catalyzes H3K27me3 promoted the CF activation and differentiation and ECM proteins deposition, which can be alleviated by EZH2 inhibitor GSK126. Therefore, EZH2 is an important factor for diabetic myocardial fibrosis.
EZH2 inhibits PPAR-γ transcription
EZH2 regulates target genes in histone methyltransferase dependent manner or in methyltransferase independent manner to suppress or co-activate transcription. After the PRC2 complex recognizes and binds the target gene, EZH2 silences target gene expression by methylation of histone H3K27 on the transcription factor of target gene [39]. In addition, EZH2 can activate downstream genes by direct methylation of non-histone proteins in a non-PRC2-dependent manner. Song et al. [30] reported that increased EZH2 promoted the development of atrial fibrillation via binding to Smad2 to form a transcription complex to increase transcription of alpha-smooth muscle actin 2 (ACTA2) and conversion of CFs to myofibroblasts with Ang II stimulation. In our study, EZH2 did not significantly increase in DM cardiac tissue, phosphorylation of EZH2 reduced with HG. EZH2 may promote the diabetic cardiac fibrosis via repress PPAR-γ in PRC2-dependent manner. PPARs, belonging to the nuclear receptor superfamily, promote or ameliorate cardiac dysfunction [40,41]. PPARs have several different isoforms, including α, β/δ, and γ. Activation of PPAR-γ with rosiglitazone ameliorates cardiac remodeling in pressure-overloaded rat hearts [22]. In activated hepatic stellate cells (HSC), H3K27me3 was accumulated on PPAR-γ promoter along with EZH2 as repressive histone marker. PPAR-γ was down-regulated in activated HSCs compared to quiescent HSCs [39]. Both pharmacological inhibition and molecular silencing of EZH2 increase PPAR-γ transcription in a way of histone methyltransferase [42]. In the present study, PPAR-γ restores transcription with GSK126 treatment compared to HG-stimulated CFs. These results revealed that EZH2 inhibited of the expression of PPAR-γ in HG-treated CFs or diabetic condition. Activation of PPAR-γ decreased viability, proliferation and myofibroblast differentiation of CFs in responses to Ang II stimuli [21]. Activation of PPAR-γ by rosiglitazone reduced TGF-β1 expression, CFs migration and proliferation, and the ECM proteins deposition. These results indicated that the EZH2/PPAR-γ pathway might be involved in HG-induced the differentiation of CFs and diabetic cardiac fibrosis.
HG regulates cardiac fibrosis through activation of AMPK/EZH2 signaling
Under persistent energy starvation, AMPK is activated by increased level of cellular adenosine-diphosphate diphosphate and adenosine monophosphate, which phosphorylates EZH2 at Thr311 [43,44]. Phosphorylation of Thr311 interrupts the interaction between EZH2 and SUZ12, resulting in reduction of histone methyltransferase activity of EZH2 and H3K27me3. Phosphorylation of EZH2 at Thr311 has been associated with increased survival in patients with ovarian and breast cancer [12]. In the present study, we discovered that phosphorylation of AMPK decreased in STZ-induced diabetic cardiac tissues and HG-treated CFs, which was consistent with the previous studies [45]. And phosphorylation of EZH2 was reduced in HG-treated CFs. While AMPK agonist up-regulated the phosphorylation of EZH2 at Thr311 and negatively regulated the activity of H3K27me3 in HG-treated CFs. These results indicated HG might inhibit phosphorylation of EZH2 via suppressing AMPK phosphorylation.
Conclusion
In the condition of HG, inactivation of AMPK led to increase of H3K27me3 by reducing the phosphorylation of EZH2 at T311, which repressed transcription of PPAR-γ and promoted diabetic fibrosis. Our data revealed AMPK/EZH2/PPAR-γ signaling pathway is involved in CFs activation, differentiation and diabetic fibrosis. Therefore, inhibition of EZH2 or activation of PPAR-γ may be a novel therapy target to ameliorate cardiac fibrosis.
Notes
CONFLICTS OF INTEREST
No potential conflict of interest relevant to this article was reported.
AUTHOR CONTRIBUTIONS
Conception or design: S.S.L., L.P., S.D.
Performed the experiments: S.S.L., L.P., Z.Y.Z., M.D.Z.
Acquisition, analysis, or interpretation of data: X.F.C., L.L.Q., M.D., Z.M.Y.
Drafting the work or revising: S.S.L., S.D., R.X.W.
Final approval of the manuscript: all authors.
FUNDING
This study was supported by grants from the National Natural Science Foundation of China (grant numbers: 81770331, 8237 0342), Natural Science Foundation of Jiangsu Province (No. BK20231145), Top Talent Support Program for young and middle-aged people of Wuxi Health Committee (grant numbers: BJ016), Program of Wuxi Translational Medicine Center (grant numbers: 2020ZHYB14).
Acknowledgements
None