Primordial Drivers of Diabetes Heart Disease: Comprehensive Insights into Insulin Resistance
Article information

Abstract
Insulin resistance has been regarded as a hallmark of diabetes heart disease (DHD). Numerous studies have shown that insulin resistance can affect blood circulation and myocardium, which indirectly cause cardiac hypertrophy and ventricular remodeling, participating in the pathogenesis of DHD. Meanwhile, hyperinsulinemia, hyperglycemia, and hyperlipidemia associated with insulin resistance can directly impair the metabolism and function of the heart. Targeting insulin resistance is a potential therapeutic strategy for the prevention of DHD. Currently, the role of insulin resistance in the pathogenic development of DHD is still under active research, as the pathological roles involved are complex and not yet fully understood, and the related therapeutic approaches are not well developed. In this review, we describe insulin resistance and add recent advances in the major pathological and physiological changes and underlying mechanisms by which insulin resistance leads to myocardial remodeling and dysfunction in the diabetic heart, including exosomal dysfunction, ferroptosis, and epigenetic factors. In addition, we discuss potential therapeutic approaches to improve insulin resistance and accelerate the development of cardiovascular protection drugs.
INTRODUCTION
Diabetes heart disease (DHD) is a structural and functional heart disease associated with diabetes mellitus (DM) and occurs in the absence of conventional cardiovascular disease (CVD). DHD mainly includes coronary artery disease, heart failure (HF), autonomic heart disease, and diabetes cardiomyopathy. Specifically, the functional features are dominated by left ventricular diastolic dysfunction, and the structural features include myocardial fibrosis, cardiac hypertrophy, and impairment of coronary microvascular perfusion [1]. Recent studies have shown that DHD is the most common and dangerous complication of DM, affecting more than half of all DM patients [2]. Despite the fact that glycemia control alone can exacerbate myocardial injury, CVD is progressing. In contrast, metformin reduces the incidence of cardiovascular damage [3]. This indicates that enhanced insulin sensitivity, rather than plasma glucose levels, plays an important role in improving DHD outcomes. Insulin resistance is the pivotal link in metabolic disorders and crucial factor in the progression of DHD [4]. Some clinical trials suggest that patients with insulin resistance are twice as likely to develop coronary heart disease as those without insulin resistance [5,6]. The incidence of CVDs in hyperinsulinemic patients exceeds three times that of non-hyperinsulinemic patients. Improving insulin resistance can reduce the risk of CVDs in DM patients by about 55% [3].
The pathophysiology of insulin resistance leading to DHD has been the topic of research since its first description. Insulin resistance can exacerbate atherosclerosis, myocardial fibrosis and ventricular hypertrophy, leading to cardiac diastolic dysfunction and eventual development of HF. The aim of this review is to provide a comprehensive and updated overview of the clinical, pathogenetic, and molecular aspects of DHD. Firstly, we introduce the pathways of insulin signaling and the development of insulin resistance. We also summarize the mechanisms of insulin resistance to DHD, including myocardial energy metabolism disorders, abnormal calcium cycling, microRNAs (miRNAs), cell cycle processes, vascular injury, cardiac autonomic dysfunction, and inflammation. Finally, we outline the potential targeted therapy for improving insulin resistance during DHD.
INSULIN RESISTANCE
Insulin resistance is a state where the body reduces insulin sensitivity and glucose absorption and processing capacity. Oxidative stress, inflammation, lipid metabolism disorders, epigenetic changes, and intestinal microorganisms can contribute to the development of insulin resistance. Although insulin responses in different cell types vary, this is largely due to different lateral effects, as all insulin-responsive cells have very similar peripheral components. To date, two major insulin signal pathways have been identified: the phosphatidylinositol 3-kinase/protein kinase B (PI3K/Akt) pathway and Ras/mitogen-activated protein kinase (RAS/MAPK) pathway (Fig. 1). The PI3K/Akt signaling pathway is the most classical insulin signaling pathway. A PI3K enzyme comprises two subunits, the catalytic subunit p110 and the regulatory subunit p85. It catalyzes the production of phosphate (3,4,5)-triphosphate (PIP3), which activates Akt by phosphoinositide-dependent kinase 1 (PDPK1) [7,8]. Phosphorylation of Akt plays an instrumental role in cell growth and proliferation, glucose and fatty acid metabolism [7,9]. Insulins also phosphorylate proteins containing SH2 structural domains (SHCs), activate RAS/MAPK signal pathways, and regulate various physiological processes such as cell growth, proliferation, differentiation, and apoptosis, without involvement in metabolism [10].

The signaling pathway of insulin action and the selective impairment of insulin receptor substrate (IRS)-protein kinase B (Akt) signaling pathway in insulin resistance. (A) It shows that insulin binding to the insulin receptor α subunit on the target cell surface activates the receptor subunit tyrosine kinase, leading to the autophosphorylation and phosphorylation of the IRS1/2, which activates the phosphatidylinositol 3-kinase (PI3K)/Akt pathway and the RAS/mitogen-activated protein kinase (MAPK) pathway. (B) It shows that in insulin-resistant state, PI3K/Akt signaling is impaired, but the MAPK pathway is preserved, which may lead to a mitogenic-promoting effect of insulin in endothelial cells involved in vascular injury. GDP, guanosine 5´-diphosphate; GTP, guanosine 5´-triphosphate; PIP3, phosphatidylinositol 3,4,5-trisphosphate; GLUT-4, glucose transporter-4; RaF, rapidly accelerated fibrosarcoma; MEK, mitogen-activated protein kinase; PDPK1, phosphoinositide dependent protein kinase 1; ASC16, 6-O-ascorbyl palmitate ester; ERK, extracellular regulated kinase; eNOS, endothelial nitric oxide synthase; NF-κB, nuclear factor-κB; FoxO1, forkhead transcription factor O1; mTOR, mammalian target of rapamycin; GSK-3β, glycogen synthase kinase-3β; FFA, free fatty acid; CD36, cluster differentiation protein 36; PPAR, peroxisome proliferator-activated receptor.
Imbalances in insulin signaling consists of two main aspects: on the one hand, inactivation of insulin receptor substrate (IRS) proteins, including down-regulation of receptor structure, number and binding affinity, and on the other hand, dysfunction of insulin signaling pathways and impairment of their signaling capacity. Any abnormal insulin signaling site can cause insulin resistance, for example, IRS degradation, phosphorylation, distribution, blockade, and odd expression of PI3K, Akt, and downstream proteins. Additionally, the effects of insulin resistance on intracellular signaling appear to be pathway-specific [11]. PI3K/Akt signaling is impaired in individuals in a state of insulin resistance, but insulin-activated MAPK pathways are often preserved and are involved in atherosclerotic (AS) lesions [12].
IMPACT OF INSULIN RESISTANCE ON DIABETES HEART DISEASE
Potential contributory mechanisms to insulin resistance-induced DHD are gradually expanding. Currently, maturation mediators identified include myocardial metabolic substrates, impaired calcium handling, miRNAs, cell cycle processes, vascular damage, autonomic nervous system of the heart, and inflammation [8,13,14]. Fig. 2 provides a more comprehensive illustration of these contributing mechanisms and the places where they may overlap.
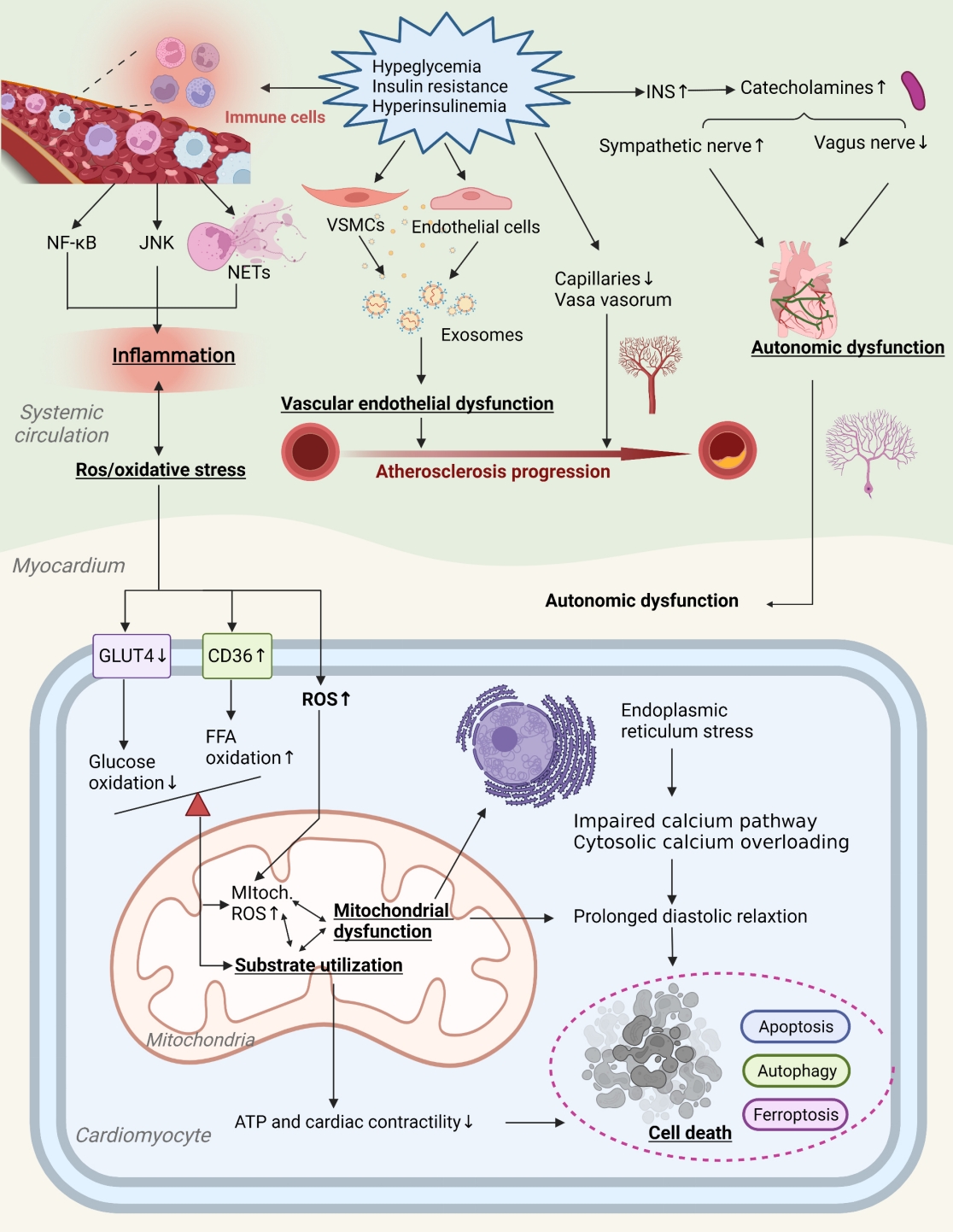
Mechanisms of insulin resistance-induced diabetes heart disease (DHD). Insulin resistance affects the progression of DHD by altering cardiovascular risk factors and reducing the insulin signaling pathway, causing a number of cardiovascular diseases such as myocardial fibrosis, ventricular hypertrophy, atherosclerosis, impaired myocardial systolic and diastolic function, and hypertension. INS, insulinase-like proteases; NF-κB, nuclear factor-κB; NET, neutrophil extracellular trap; VSMC, vascular smooth muscle cells; GLUT-4, glucose transporter-4; CD36, cluster differentiation protein 36; FFA, free fatty acid; ROS, reactive oxygen species; ATP, adenosine triphosphate.
Insulin resistance affects DHD through myocardial metabolic substrates
The heart derives approximately two-thirds of its energy requirements from fatty acid oxidation and one-third from glucose oxidation, maintaining a relative balance between the two. As DHD advances, cardiac energy substrate metabolism is altered: cardiomyocytes shift from using both glucose and fatty acid oxidation to relying almost exclusively on fatty acid β-oxidation for energy, resulting in impaired synthesis of high-energy phosphate compounds in cardiac muscle tissue [15]. Insulin resistance plays a role in this change. Depending on the daytime indoor lighting environment, insulin-resistant patients affect the metabolism of the body’s substrate and energy consumption in a time-dependent manner, including an increase in fatty acid oxidation, a reduction in glucose oxidation and glycolysis [16].
Free fatty acid
Free fatty acid (FFA) is the primary energy source of the myocardium. Increased expression of peroxisome proliferator-activated receptor α (PPARα) in cardiomyocytes from diabetic patients is associated with intake of FFA, accumulation of triacylglycerol and reduced use of glucose [15]. Furthermore, increased FFA release from fat tissue in turn affects FFA transport in heart cells. Cluster differentiation protein 36 (CD36) promotes the absorption of FFA in cardiomyocytes and increases cardiac lipotoxicity. It was found that insulin resistance induces CD36 upregulation in H9C2 cells and promotes inflammation and lipid accumulation, exacerbating diabetic myocardial metabolic disorders [17,18]. An AMP-activated kinase (AMPK) is an energy-sensing enzyme that is compensatively increased during low cellular energy levels [15]. Insulin resistance status stimulates AMPK, which increases FFA uptake by the heart via Fat -CD36 and increases DHD burden [19]. Abnormalities in lipid metabolism tend to accelerate the development of DHD. Insulin resistance makes insulin ineffective in inhibiting adipocyte lipolysis and increase FFA in the liver, which further stimulates the accumulation and secretion of low-density lipoproteins, leading to disorders of lipid metabolism, lipotoxicity, cardiac steatosis and even myocardial cell necrosis, resulting in diabetic myocardial damage [20].
Glucose
The oxidation of glucose and pyruvate is an important factor in the formation and storage of myocardial energy. Pyruvate dehydrogenase (PDH) is an important enzyme that regulates the balance of heart carbohydrates and lipid metabolism. In patients with DHD, PDH activity is reduced and pyruvate oxidation is impaired, which seems to be associated with insulin resistance [21]. The dissociation of glycolysis and pyruvate oxidation in the diabetic heart leads to the accumulation of glycolytic intermediates, which activates specific glucose-sensitive transcription factors. Glucose enters cardiomyocytes through glucose transporters 1 and 4, where GLUT1 is located on the cell membrane, and GLUT4 is transferred to the cell membrane under the activation of Akt, and regulates genes such as glycogen synthase kinase-3 (GSK-3) and forkhead box protein O1 (FoxO1) which promote glucose uptake in cardiomyocytes and inhibit glycogen synthesis [22]. Under conditions of hyperglycemia and insulin resistance, the PI3K/Akt signaling pathway that stimulates the GLUT4 recruitment to plasma membrane is impaired, causing the production of advanced glycation end products (AGEs) and the activation of the chronic hexosamine biosynthesis pathway, leading to heart glucotoxicity and myocardial dysfunction [23,24].
Ketone bodies
Ketone bodies are easily oxidized by the heart. Therefore, when the metabolism of other energy substrates is dilated, the ketone body may represent an additional source of energy for the diabetic heart. In fact, increased regulation of enzymes involved in ketone metabolism has been reported in both patients with DHD and advanced HF mice models. Increasing circulating ketone bodies by intravenous infusion of ketone bodies significantly improves cardiac function in patients with DHD [25]. A ketogenic diet extends the lifespan in mice with insulin resistance and improves memory in aged mice [26,27]. In addition, some researchers found that the concentration of ketone bodies was negatively correlated with insulin resistance and triglycerides [28]. Consequently, insulin resistance can affect energy metabolism in the diabetic heart by regulating ketones metabolism.
Insulin resistance affects DHD through impaired calcium handling
In cardiomyocytes, the calcium cycle maintains physiological excitation-contraction couplings. The sarcoplasmic reticulum (SR) is the main site of the cardiac calcium cycle. Many Ca2+ enter the cardiomyocytes through L-type calcium channels, induces SR calcium release through the ryanodine receptor (Ryr), and regulates myocardial contractility. Additionally, the SR reuptake of cardiomyocyte Ca2+ is mediated by increased activity of the sarcoplasmic reticulum Ca2+ pump (SERCA), the Na+/Ca2+ exchanger (NCX), and the Ca2+ATPase [23,29]. In diabetes cardiomyopathy, abnormal expressions or activities of Ryr receptors, SERCA2a and NCX allows impaired Ca2+ transients as well as Ca2+ uptake by the SR, affecting myocardial contractile function. At the same time, impaired Ca2 efflux from the extracellular matrix, impaired relaxation of cardiomyocytes, and sustained increase in action potential time can lead to diastolic dysfunction [15,23,30]. These data indicate that weakened Ca2+ treatment in the cardiovascular cells plays an important role in the development of diastolic dysfunction characteristics of early diabetes cardiomyopathy.
Insulin can enhance the function of the calcium channel through the PI3K/Akt signaling pathway, activate SERCA2a and improve myocardial contraction [8]. On the other hand, a state of cardiac insulin resistance attenuates PI3K/Akt signaling, and decreased glucose uptake can reduce Ca2+ ATPase activity, making it challenging to move Ca2+ back into the SR, resulting in impaired calcium handling [23,29]. A study finds that heart insulin resistance can impair Ca2+ homeostasis through the protein kinase B/striated muscle preferentially expressed protein kinase/SERCA2a (PKB/SPEG/SERCA2a) pathway and promote the development of diabetes cardiomyopathy [31]. The state of insulin resistance also significantly promotes calcium ion and calcium/calmodulin-dependent protein kinase II phosphorylation, thereby affecting calcium metabolism [32]. In addition, reduced nitric oxide (NO) bioavailability can affect T-type Ca2+ channels through the cyclic guanosine monophosphate/protein kinase G (cGMP/PKG) signaling pathway, aggravate myocardial fibrosis caused by hyperglycemia, and lead to contraction deformation of myocytes [33,34].
Insulin resistance affects DHD through MiRNAs
MiRNAs are small single-stranded RNA molecules of the noncoding RNA family, which interfere with the repression of mRNA and degradation of protein production by target genes at the posttranscriptional level.
Diabetes cardiomyopathy is highly correlated with dysregulated expression of miRNAs. Some miRNAs, such as miR-1 and miR-133a, which are abundantly expressed in cardiomyocytes, are significantly reduced in DM patients, thus contributing to the development of diabetes cardiomyopathy [35]. In streptozotocin-induced DM rats, the overexpression of miR133a improved myocardial contractility and mitigated myocardial injury by upregulating tyrosine aminotransferase [36]. In addition, insulin resistance affects the expression levels of miR-155-5p and miR-143-3p, exacerbating diabetes-related vascular disease. MiR-155-5p was overexpressed and miR-143-3p was weakened in atherosclerosis in mice and humans, causing plaque instability and accelerating the progression of atherosclerosis [37]. Insulin resistance is clearly responsible for affecting epigenetics and contributing to DHD development. MiR-690 is an exosome-derived miRNA from M2-polarized macrophages. Recent studies have shown that miR-690 improves glucose tolerance and insulin in obese mice, and it can be a novel insulin sensitizer for metabolic heart disease [38].
Insulin resistance affects DHD through cell cycle processes
Mature cardiomyocytes are non-regenerative. Cell death is the main form of cardiomyocyte reduction and is a fundamental cause of cardiac hypertrophy, myocardial fibrosis and ventricular remodeling, leading to progressive HF.
Apoptosis
Apoptosis is a programmatic death process regulated by various genes and proteases. The researchers found that elevated levels of apoptosis markers such as TNF receptor 1 (TNFR-1), TRAIL receptor 2 (TRAILR-2), and Fas increased the risk of myocardial infarction and stroke in patients with DHD [39]. In the PI3K/Akt signaling pathway, in addition to the phosphorylation of mammalian target of rapamycin (mTOR) protein, the GSK-3β/β-catenin signaling pathway can also improve cardiomyocyte apoptosis and improve myocardial injury in rats with myocardial ischemia-reperfusion [40,41]. However, in insulin resistance, the PI3K/Akt signaling pathway is inhibited, but the RAS/MAPK signaling pathway still functions, inducing apoptosis and necrosis in cardiac myocytes and accelerating the progression of diabetes cardiomyopathy [42]. It has been confirmed that the apoptosis of cardiomyocytes induced by insulin resistance depends on dose [43]. In addition, lipotoxicity due to insulin resistance may also lead to increased apoptosis [44]. Endoplasmic reticulum stress (ERS) is involved in apoptosis. In mice fed a high-fat diet, down-regulating ERSrelated proteins and inhibiting the pathways that regulate ERS and sterol regulatory element-binding protein (SREBP)-1c/SREBP-2 improved insulin resistance [45]. Therefore, insulin resistance can play an important role in activating the apoptosis pathway of the ERS cell, which could become an essential path for studying the DHD apoptosis mechanism.
Autophagy
Autophagy is a highly preserved catabolic process, which plays a key role in maintaining cell homeostasis by degrading and recycling the cytoplasmic content, but this process is often impaired in diabetic hearts [46]. In preclinical tests, dysregulation of autophagy was observed in the diabetic heart [47]. Insulin is an essential regulator of myocardial autophagy, and the unc-51-like kinase 1 (ULK1) dot is an early marker of autophagy formation. It was found that insulin can regulate autophagy by altering ULK1 phosphorylation and FoxO1/3 activation through the Akt/mTOR signaling pathway [48]. Insulin resistance exacerbates diabetes-related cardiac injury by affecting ULK1 expression and altering the expression levels of p62 and light chain 3I (LC3I)/LC3II proteins, leading to dysregulation of cardiomyocyte autophagy [49]. The mTOR mediates autophagy inhibition, and its inhibitor rapamycin improves insulin resistance, alleviates glucose and lipid metabolism disorders, reduces inflammatory reactions, and promotes autophagy [50]. Thus, rapamycin may be a potential treatment for diabetes-related myocardial autophagy. In addition, autophagy can also adversely affect insulin resistance, and a lack of the autophagy related 16-like 1 (ATG16L1) autophagy gene can cause insulin resistance by destroying insulin receptor substrates-1 (IRS1) through Kelch-like 9/Kelchlike 13/complex with cullin 3 (KLHL9/KLHL13/CUL3) mechanisms [51]. Visceral adipose tissue-derived serine protease inhibitor (vaspin) reduces insulin resistance, metabolic disorders and hepatic steatosis [52]. Vaspin was found to protect against myocardial ischemia/reperfusion by upregulating the autophagy flux dependent on AMPK-mTOR and the recovery of lysosomal functions [53]. Vaspin also increased the level of myocardial autophagy and decreased the rate of apoptosis and fibrosis of cardiomyocytes in diabetic rats [54]. Vaspin may be a potential target of autophagy in DHD patients.
Ferroptosis
Ferroptosis is caused by the accumulation of lipid peroxides dependent on iron. A study found that the ferroptosis is an important pathogen for the development of diabetes cardiomyopathy, using transmission electron microscopy to study the expression of key regulated ferroptosis in mice, and to observe typical morphological changes in ferroptosis in cardiomyocytes [55]. Glutathione (GSH) and glutathione peroxidase 4 (GPX4) are involved in ferroptosis. Targeting the guanine-rich sequence binding factor 1 (Grsf1)/GPX4 axis improves insulin resistance, inhibits oxidative stress and iron reduction, and protects damaged myocardium [56,57]. GSH formation is dependent on solute carrier family 7 member 11 (SLC7A11), which is an important molecule for the mitigation of lipid peroxidation and ferroptosis. In the hearts of mice with insulin resistance, SLC7A11 and GSH levels were significantly downregulated, leading to ferroptosis in cardiomyocytes [58]. These confirm that insulin resistance disrupts normal cellular function and enhances lipid peroxidation, leading to iron overload in cardiomyocytes and participating in the progression of DHD mechanisms. Nuclear factor erythroid 2-related factor 2 (Nrf2) plays an important role in maintaining the cell redox through the regulation of multiple antioxidants. Recent studies suggest that activation of Nrf2 to inhibit ferroptosis can be a potential therapeutic target for DHD [59]. However, it is not yet clear how Nrf2 activation alters DHD pathogenesis and ferroptosis during development.
In addition to myocardial cell death pathways, insulin resistance hampers the ability of adult heart stem cells and progenitor cells to expand and differentiate, thereby further deteriorating their senescence phenomena and accelerating the development of DHD [60,61]. Eliminating adipocytes in mice with high-fat diets improves insulin sensitivity, inflammation and glycemic status [62]. It is clear that the senescent cells accumulate in the process of insulin resistance that causes DHD. Another feature of senescent cells is the activation of the senescence-associated secretory phenotype (SASP). Removal of senescent cells using senolytics could decrease cardiac fibrosis and hypertrophy [63]. Therefore, the removal of senolytic-mediated senescent cells can be a means of improving insulin resistance, reducing cardiac remodeling, and preventing DHD progression.
Insulin resistance affects DHD through vascular damage
Vascular damage is a major link between insulin resistance and DHD. Vascular endothelial dysfunction (VED), including reduced bioavailability of NO, higher intracellular adhesion molecule-1 (ICAM-1) and endothelin-1 (ET-1), results in reduced vasodilator activity and instability of lipid plaques, which severely affects vascular function and structure. Insulin resistance is positively correlated with VED and can exacerbate vascular damage related to diabetes [64]. In insulin-resistant states, aldosterone and insulin activate glucocorticoid kinase1intheserum by the corticoid salt receptor and insulin receptor, triggering synergistic activity of the sodium channel of the endothelial vascular endothelium, reducing NO production and exacerbating vascular sclerosis [65]. By modeling the dynamics of insulin signaling in the vascular endothelium, insulin resistance is further confirmed to affect PI3K/NO and MAPK/ET-1 pathways, thereby inhibiting endothelial nitric oxide synthase (eNOS) and increasing ET-1 and adhesion molecules [66].
Exosomes
Exosomes are nano-scale vesicles released by cells. They consist of a lipid bilayer and contain several biomolecules that act as intercellular communication by focusing on receptor cells to release their content. Exosomes secreted by endothelial cells and vascular smooth muscle cells (VSMC) are important in maintaining vessel wall homeostasis. Exosomes secreted by oxidized LDL-treated human arteries (human umbilical vein endothelial cells [HUVECs]) cause hyperlipidemia and increase the expression of the transcripts of metastatic lung adenocarcinoma 1, which increases local inflammation and causes the formation and progression of AS plaques [67]. The exosome plays a role in the mechanisms of insulin resistance causing DHD. The expression of arginase 1 in serum exosomes of diabetic mice and diabetic patients increased, and it was found that it was captured by endothelial cells, inhibiting endothelial NO production and thus promoting DHD-related endothelial functional impairment [68]. Furthermore, circRNA-0077930, delivered by HUVEC exosomes in insulin-resistant states, induces VSMC senescence by lowering the expression of miR-622 and regulating Kras, p21, p53, and p16, which can accelerate the pathological progression of diabetic vascular dysfunction [69]. Exosomes are clearly involved in pathology of vascular damage associated with insulin resistance.
Endothelial progenitor cells
An endogenous repair process can alleviate vascular endothelial injury. Circulated endothelial progenitor cells (EPCs) are mediators of endothelial repair and contribute to angiogenesis and functional recovery in ischemic tissues in DHD patients [70,71]. Insulin resistance can disrupt EPC function and increase cardiovascular and metabolic risk [71]. The number and location of EPCs is inversely linked to insulin resistance [72]. The use of insulin sensitive metformin and rosiglitazone inhibitors prevents the activation of nuclear factor-κB (NF-κB) and inflammation in the local concentration of PM2.5, improves insulin resistance, and restores the circulating EPC level [73]. Additionally, Nrf2 regulates the survival and angiogenesis functions of EPC. It was found that the expression level of Nrf2 and its downstream genes decreased in the EPCs of DHD patients and db/db mice, and that the increase in the expression of NRF2 increased the resistance of EPCs to the oxidative damage caused by diabetes [74]. Therefore, Nrf2 is a potential therapeutic target to improve insulin resistance, restore EPC function, and inhibit the progression of DHD.
Vasa vasorum
Vasa vasorum (VV) surrounds the middle and outer layers of the walls of large and medium blood vessels, providing oxygen and nutrients to the walls of blood vessels and surrounding tissues, and expelling cellular metabolic waste. Patients with DHD have impaired VV, resulting in coronary microcirculation disturbances and consequently impaired coronary flow reserve. Several researchers have analyzed cardiac capillary density in patients with end-stage HF and found reduced capillary diameter and higher vascular permeability in patients with combined DM [75]. Insulin resistance states can damage the VV and rare microvasculature, thereby causing vasospasm [76]. Studies have shown that in insulin-resistant environments, factors such as tumor necrosis factor (TNF) and vascular endothelial growth factor (VEGF) can impair vascular maturation and result in smaller, weaker, potentially fracturing vessels, thrombosis and cardiovascular events [77,78]. Another study showed that insulin resistance adipocyte-derived exosomes (IRADEs) exacerbated the insulin resistance status of diabetic ApoE-/- mice, promoted VV production, and increased plaque burden and AS plaque susceptibility index [79]. More research is still needed to demonstrate the relationship between VV and diabetic vascular dysfunction.
Insulin resistance affects DHD through cardiac autonomic nerves
Cardiac adipose tissue possesses rich neural distribution. Autonomic dysregulation in patients with DHD may induce arrhythmias, such as ventricular fibrillation and atrial fibrillation. Insulin resistance is closely related to the function of the autonomous nervous system. This was confirmed in a study by using electrophysiological methods to observe autonomic tone in rats fed a high-fat diet [80]. Heart rate variability (HRV) is currently the most common non-invasive index for evaluating autonomic function. Patients with insulin resistance have impaired cardiac autonomic function and reduced HRV indicators [81]. The reason may be that insulin resistance increases the level of insulin and the abnormal increase in catecholamine levels, which affects the four phase depolarization of the heart and electrical activity of the heart, causing the decrease in vagal activity and the increase in sympathetic activity.
The nerve growth factor (NGF) is a nutritional factor for sympathetic nerve survival and is vital for sympathetic nerve damage repair. Starting from day 3 after myocardial infarction, the level of intramyocardial NGF increased significantly, and the level of sympathetic regeneration was consistent with the level of intramyocardial NGF. NGF reduces insulin resistance, activates IRS1, and improves glucose metabolism and c-Fos expression [82]. It was shown that NGF levels in diabetic mice have decreased and sirtuin 1 (SIRT1) and NGF expression in neuronal tissues have increased, protecting diabetic animals from neuronal damage caused by hyperglycemia [83]. Purinergic ligand-gated ion channel 3 (P2X3) receptors are mainly expressed in primary sensory neurons and are associated with various pathological pains, especially inflammatory pain. P2X3 was found to be involved in the desensitization of afferent neurons in chronic HF and the expression of P2X3 receptors in the L4/5 dorsal root ganglion of rats with HF was 2.8-fold higher than in controls [84]. P2X3 shRNA can reduce sympathetic activity by interfering with P2X3 receptor expression in stellate ganglia, thereby alleviating diabetic cardiac autonomic neuropathy [85]. In addition, a study has found that hyperglycemia can be involved in autonomic injury in DM patients by regulating the activation of the metabolism and/or oxidative states of neuronal cells, such as increased production of the polyol pathway, hexosamine pathway, and AGE [86]. Additionally, renin-angiotensin-aldosterone system (RAAS) activity in the paraventricular nucleus of the hypothalamus raises the signaling of extracellular signal-regulated kinase 1/2 (ERK1/2) MAPK and plays an important role in mediating the sympathetic movement of HF mice [87]. These studies initially suggest that in insulin resistance, autonomic dysfunction is an important part of diabetic cardiac autonomic neuropathy.
Insulin resistance affects DHD through inflammation
The role of inflammation in insulin resistance-induced CVDs has been investigated. DHD is often accompanied by high levels of circulating proinflammatory cytokines, such as TNFα, interleukin 6 (IL-6), and IL-18, and cytokine receptors [88]. Insulin resistance is a chronic low-level inflammation state that activates the NF-κB in flammatory pathway, induces NLR family pyrin domain containing 3 (NLRP3) inflammasome expression, and improves chronic inflammatory responses in the system [89]. Furthermore, activation of the NLRP3 inflammasome can exacerbate caspase-1 recruitment, promote cystatin-1 activation, and cleave IL-1β and IL-18 precursors, thereby enhancing the proinflammatory cascade [90,91]. In the type 2 diabetes mellitus (T2DM) rat model, activated NLRP3 inflammasome leads to inflammatory cell expansion and infiltration, which plays a crucial role in the pathogenesis of HF, and reduced NLRP3 inflammasome causes cardiomyopathy in T2DM rats [92]. Moreover, insulin resistance can also induce the c-Jun N-terminal kinases (JNKs) inflammatory pathway.
An important feature of the inflammatory response is the migration and aggregation of neutrophils. NETosis is a specific neutrophil cell death process in which neutrons release their nucleiin the form of neutron extracellular traps (NETs) [93]. NETosis plays a role in insulin resistance-induced inflammatory heart disease, including myocardial injury and vascular disease. An animal model established by Wnt5a-mediated infiltration of neutrophils into the heart confirmed that NETs lead to excessive inflammation and cardiac insufficiency [94]. Another study has also confirmed the potential role of excess or dysregulated NETosis in atherosclerosis development [95]. NETs release products are significantly increased in insulin-resistant patients [96]. Therefore, NETs may mediate the inflammatory response between insulin resistance and DHD. Furthermore, insulin resistance leads to a switch from a proinflammatory M2 to an anti-inflammatory M1 macrophage polarity, thereby escalating inflammatory responses [91]. Early anti-inflammatory or insulin sensitization treatment strategies have potential benefits in individuals with features of insulin resistance syndrome.
In the state of insulin resistance, reactive oxygen species increased dramatically, and endogenous antioxidant factors such as superoxide dismutase (SOD) and glutathione peroxidase (GSH-Px) were decreased in cardiomyocytes, exacerbating the high oxidative stress state [97]. The family protein nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 1 (Nox1) is involved in the oxidative stress process. This protein is up-regulated in diabetic mice, and knocking down it can delay the progression of atherosclerosis [98]. Therefore, Nox1 could be a potential target for improving insulin resistance and delaying the progression of DHD.
TARGETING INSULIN RESISTANCE IN DHD
Here, we mainly aimed to identify agents that both improve insulin resistance and have cardiovascular benefits. These agents can directly or indirectly target IRS1 and IRS2 and their associated protein kinases and gene expression to activate insulin signaling pathways, which may provide potential therapeutic options for the prevention and treatment of insulin resistance and its cardiovascular complications (Table 1).
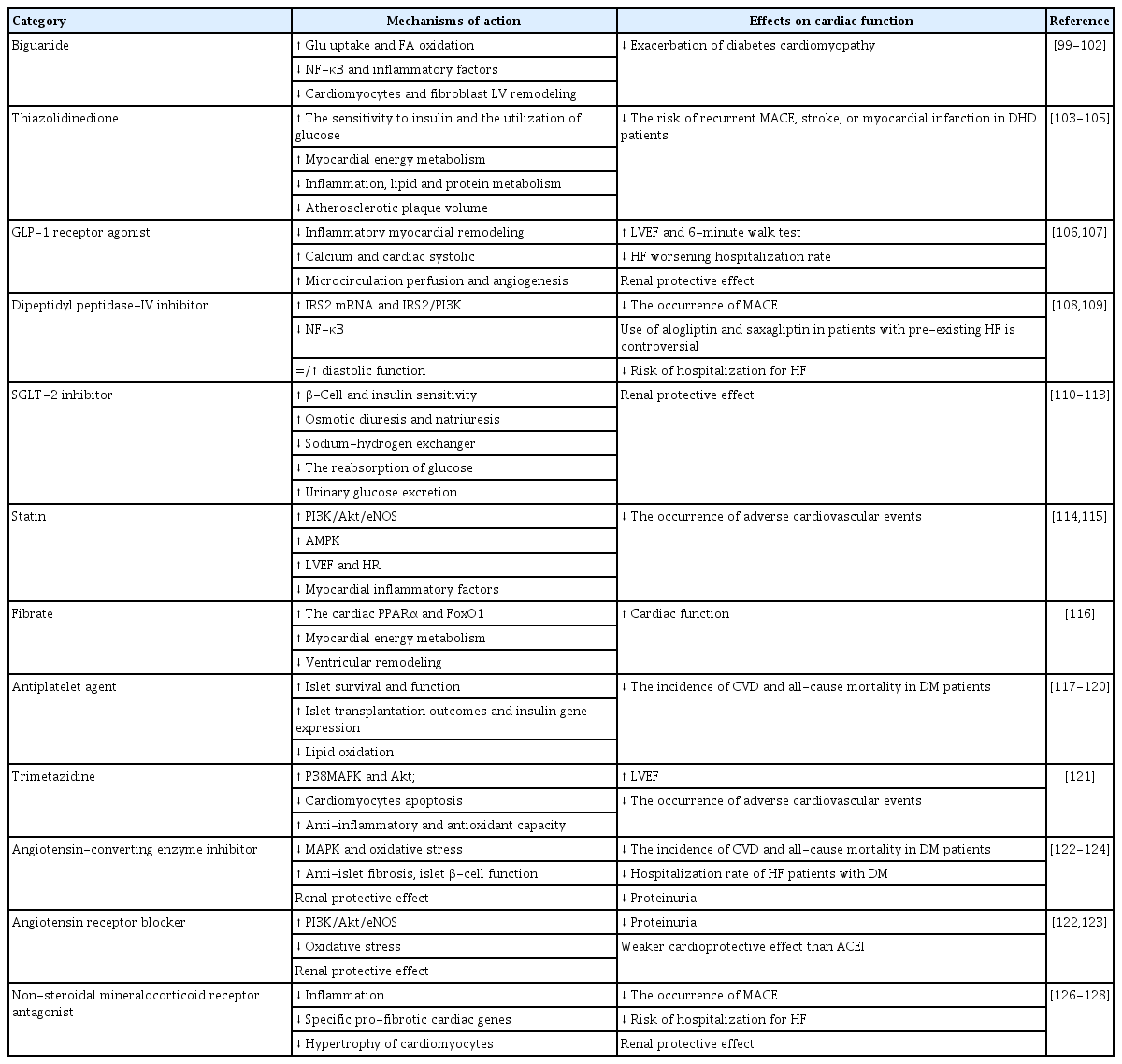
A review of the main efficacy of different drugs for insulin resistance and DHD
Hypoglycemic agents are currently the first choice of drugs for clinical improvement of insulin resistance. Biguanides is currently the first-line drug for the treatment of DM with HF [99-101]. A 49-year prospective open randomized study of patients with metabolic syndrome and diabetes showed that the use of metformin (1,000 mg two times a day) in addition to an improvement in lifestyle significantly reduced insulin resistance and improved diabetes [102]. Thiazolidinediones are PPARγ agonists that increase tissue sensitivity to insulin, enhance glucose utilization, and reduce blood FFA concentrations to improve myocardial energy metabolism [103-105]. However, edema is a potential side effect and should be used with caution in subjects at risk for HF, and the degree of edema should be monitored regularly. Exenatide, a glucagon-like peptide 1 receptor (GLP-1R) agonist, improves calcium circulation, cardiac systolic function and endothelial function, and has the potential to treat T2DM combined with HF [106,107]. However, this class of drugs may directly affect sinus node myocytes and enhance sympathetic nervous system activity, and the therapeutic effect in HF clinical treatment is unstable. Dipeptide-IV inhibitors inhibit the MAPK and NF-κB signal pathways, reduce inflammatory responses and improve insulin resistance [108]. However, in the EXAMINE trial, alogliptin increased the incidence of HF in patients who had signs of HF at the time of randomization grouping [109]. Further evidence is needed to draw reliable conclusions about the cardiovascular safety of alogliptin in patients with T2DM. Sodium glucose co-transporter 2 inhibitor (SGLT-2i) are novel glucose-lowering agents that promise to break the vicious cycle of insulin resistance and HF. Englestrin improves β-cell function and insulin sensitivity, regulates mitochondrial function, reduces plasma volume and osmotic diuresis, attenuates ventricular remodeling, myocardial energy metabolism, and adipokine kinetics, and mediates cardioprotective and renoprotective effects [110-113]. Further clinical trials are needed to verify long-term safety and effectiveness.
Some lipid lowering agents have shown have shown numerous effects beyond the ability to control blood lipids and improved insulin resistance. Statins can increase AMPK activation and inhibit FoxO1 to counteract cardiac hypertrophy. However, some studies have found that statins can cause insulin resistance, possibly due to differences in statin doses [114,115]. Fibrates downregulates phosphokinase and phosphatase-1 activity in mitochondria and improves insulin sensitivity [116]. However, such drugs have the disadvantages of low bioavailability, poor patient compliance, and individual differences. There is no clinical study of fibrates in the treatment of T2DM complicated by HF. Antiplatelet agents help to improve islet survival and function, islet graft outcome, and insulin gene expression [117-120]. However, platelet reactivity is enhanced in patients with insulin resistance, and the risk of thrombosis and bleeding should be taken into account. Trimetazidine is an anti-anginal drug. Studies have shown that trimetazidine activates insulin signaling pathways, reduces cardiomyocyte apoptosis and enhances anti-inflammatory and antioxidant capacity, significantly improving glycated hemoglobin, blood glucose, myocardial velocity and other indices [121]. RAAS inhibitors RAAS inhibitors can resist islet fibrosis, improve islet β-cell function and protect the kidney [122]. Early intervention can prevent the development of microalbuminuria in patients with T2DM. Angiotensin-converting enzyme inhibitors (ACEIs) have a more advantageous cardioprotective effect than angiotensin receptor blockers (ARBs) [123]. New compounds of angiotens inreceptor enkephalinase inhibitors (ARNis) improve cardiac function, promote energy expenditure, and reduce hospitalization rates of HF patients with DM [124,125].
Recent clinical trials have demonstrated the benefits of non-steroidal mineralocorticoid receptor antagonists (MRAs) in DM patients. Finerenone, a non-steroidal highly selective MRA, was developed based on the structure of dihydropyridine [126]. The secondary analysis of the FIGARO-DKD Trial on HF has confirmed that finerenone can significantly reduce the incidence of new HF, improve HF-related prognosis, and reduce cardiovascular death or the composite endpoint of hospitalization for HF, for patients with T2DM and chronic kidney disease, regardless of their history of HF [127]. The HF subgroup analysis of the FIDELITY-DKD Trial supports this statement [128]. Finerenone brings a new therapeutic option for the prevention and treatment of cardiovascular risk management in patients with DHD. α-1 Adrenergic receptor antagonists can be used alone or in combination with other drug groups in a broad range of hypertensive patients. They can also improve insulin sensitivity and adverse blood lipids in many hypertensive patients [129,130]. β-Blockers are crucial to the treatment of patients with HF with reduced ejection fraction [131]. Currently, the evidence that selective α/β blockers improve insulin sensitivity is still very minimal, and more research is needed to verify this.
In addition to the above agents, some targets strengthen the link between insulin resistance and CVD, such as O-GlcNAcase (OGA), SPEG, fibroblast growth factor 2 (FGF2), and exosomes like vesicles (ELVs). Researchers have discovered that DM patients are also at risk for heart disease from the hyperglycemia-related hexosamine biosynthesis pathway and the OGlcNAcylation pathway. Chronic elevation of O-GlcNAcylation induces mitochondrial dysfunction and impaired left ventricular function [132]. Moreover, reducing excess O-GlcNAcylation is beneficial for restoring Ca2+ handling and cardiac contractile function in cardiomyocytes [133]. To prevent diabetes cardiomyopathy, targeting hexosamine biosynthesis or O-GlcNAcylation may be a promising approach. SPEG, a member of the myosin light chain kinase family, can regulate SERCA2a activation and is a new target for treating diabetes cardiomyopathy [132,134]. In addition to these new targets, FGF2 produced by skeletal muscle has similar effects to FGF1 in adipocytes, both promoting lipolysis and acting through FGF receptors. Studies have found that FGF2 can work directly on cardiomyocytes to maintain myocardial integrity and function and prevent damage during oxidative stress [135], which may serve as a potential target for insulin sensitization and cardiovascular protection. In addition, ELV dysregulation plays a functional role in various metabolic, autoimmune, and CVDs. ELV quantity is positively correlated with insulin resistance, and macrophages treated with ELV can inhibit the phosphorylation of Akt in human adipocytes [136,137]. Targeting ELV may be a novel mediator and therapeutic target for treating insulin resistance and β-cell exhaustion.
CONCLUSION AND PERSPECTIVE
Insulin resistance and subsequent metabolic disorders are the main drivers of the pathological processes specific to DHD. Although many current studies are underway, the pathology of insulin resistance, which affects the structure and function of the heart, has not yet been fully explained, including mitochondrial dysfunction, amino acid transport and metabolism, additional signaling molecules, hormones, kinases, phosphatases, and modifications of gene expression [138]. It was found that the oxidative capacity of mitochondria in T2DM mice was reduced by dysregulating mitochondrial fission/fusion proteins, including dynamin-related protein-1 (DRP1) and optic atrophy-1 (OPA1) [56,139,140]. Insulin resistance is involved in the dysfunction of the mitochondria in the heart through the AktmTOR-NF-κB signaling pathway, resulting in a reduction in the potential of the mitochondrial membrane, a decrease in the activity of the electron transport chain, and abnormal mitochondrial biogenesis [8]. In the future, as a research methodology, different signaling pathways of physiological effects of insulin can be used as mainline modules, and the interrelationships between each module and the upstream and downstream of different pathways can be analyzed to further clarify the specific pathological mechanisms of insulin resistance to DHD. In addition, the link between intestine dysbiosis and insulin resistance has gained attention [141]. Researchers found that the intake of probiotics in rats significantly improved insulin resistance caused by high sugar diets, reduced low-level chronic inflammation and oxidative stress, and regulated the body’s energy metabolism [142]. Intestinal flora imbalance is a breakthrough in the study of insulin resistance and the DHD mechanisms. In addition to medication, regular exercise is an important modifiable factor in improving cardiac function in patients with DHD. A nationwide retrospective cohort study in Korea found an association between sustained active physical exercise and a reduced risk of major adverse cardiovascular events in patients with DM, regardless of the type or amount of exercise [143].
Since subclinical cardiac disorders can be reversible when detected early, insulin replacement therapy and insulin sensitive chemotherapy are essential to reduce the risk of HF in DHD patients. Future urgent research is needed to rigorously test this possibility. Presently, there are still no ideal clinical interventions to combat insulin resistance in childhood [144,145]. Careful stratification or phenotyping of subjects may help to identify which signaling pathways are disturbed and facilitate the development of target-specific therapies. In addition, exosomes as a breakthrough with corresponding inhibitors will be a new target for the prevention or treatment of insulin resistance and DHD.
Notes
CONFLICTS OF INTEREST
No potential conflict of interest relevant to this article was reported.
FUNDING
This work was supported by the “Hundred Million” Talent Project of Chinese Medicine Inheritance and Innovation (QI Huang Project) QI Huang Scholar (Zhang Junping) Special Grant (No. 2021); Tianjin Famous Chinese Medicine Doctor (Zhang Junping) Inheritance Studio Special Grant (No. 2020); National Natural Science Foundation of China Project (30672734).
Acknowledgements
None